![[alpha]](../greek/ALPHA.GIF) -addition (alpha-addition); -effect; -elimination
-addition (alpha-addition); -effect; -elimination
Contents
abstraction; acceptor number (AN); acid; acidity; acidity function; activated complex; activation energy; addend; addition; addition reaction; additivity principle; adduct; A-factor; agostic; alcoholysis; allylic substitution reaction; alternant; ambident; amphiphilic; amphiprotic (solvent); amphoteric; anchimeric assistance; anionotropic rearrangement (or anionotropy); anion radical; annelation; annulation; annulene; antarafacial, suprafacial; anti; antiaromatic; anti-Hammond effect; anti-Markownikoff addition; aprotic (solvent); aquation; aromatic, aromaticity; Arrhenius equation; aryne; association; asymmetric induction; atomic orbital; attachment; autocatalytic reaction; automerization; autoprotolysis; autoprotolysis constant; "A" value; + azacarbene; + azene; + azylene; -addition (alpha-addition); -effect; -elimination
A chemical reaction or transformation, the main feature of which is the bimolecular removal of an atom (neutral or charged) from a molecular entity. For example:
(proton abstraction from acetone)
(hydrogen abstraction from methane)
See detachment.
A quantitative measure, devised by GUTMANN (1976), of Lewis acidity.
A molecular entity or chemical species capable of donating a hydron (proton) (see Brønsted acid) or capable of forming a covalent bond with an electron pair (see Lewis acid). See also hard acid.
(1) Of a compound:
For Brønsted acids it means the tendency of a compound to act as a hydron donor. It can be quantitatively expressed by the acid dissociation constant of the compound in water or some other specified medium. For Lewis acids it relates to the association constants of Lewis adducts and
(2) Of a medium:
The use of the term is mainly restricted to a medium containing Brønsted acids, where it means the tendency of the medium to hydronate a specific reference base. It is quantitatively expressed by the appropriate acidity function.
Any function that measures the thermodynamic hydron-donating or -accepting ability of a solvent system, or a closely related thermodynamic property, such as the tendency of the lyate ion of the solvent system to form Lewis adducts. (The term "basicity function" is not in common use in connection with basic solutions.) Acidity functions are not unique properties of the solvent system alone, but depend on the solute (or family of closely related solutes) with respect to which the thermodynamic tendency is measured.
Commonly used acidity functions refer to concentrated acidic or basic solutions. Acidity functions are usually established over a range of composition of such a system by UV/VIS spectrophotometric or NMR measurements of the degree of hydronation (protonation or Lewis adduct formation) for the members of a series of structurally similar indicator bases (or acids) of different strength: the best known of these functions is the Hammett acidity function Ho (for uncharged indicator bases that are primary aromatic amines). For detailed information on other acidity functions, on the evaluation of acidity functions, and on the limitations of the concept, see ROCHESTER (1970), COX and YATES (1983), HAMMETT (1940, 1970).
An activated complex, often characterized by the superscript
See energy of activation.
See addition reaction.
(1) Refers to addition reaction or addition transformation.
(2) Loosely, the formation of an adduct. (For an example, see Lewis acid.)
(3) Loosely, any association or attachment.
A chemical reaction of two or more reacting molecular entities, resulting in a single reaction product containing all atoms of all components, with formation of two chemical bonds and a net reduction in bond multiplicity in at least one of the reactants. The reverse process is called an elimination reaction. The addition may occur at only one site (
(b) Br2 + CH2=CH-CH=CH2
If the reagent or the source of the addends of an addition are not specified, then it is called an addition transformation. See also addition,
The hypothesis that each of several structural features of a molecular entity makes a separate and additive contribution to a property of the substance concerned. More specifically, it is the hypothesis that each of the several substituent groups in a parent molecule makes a separate and additive contribution to the standard Gibbs energy change (or Gibbs energy of activation) corresponding to a particular equilibrium (or rate of reaction). For further information and examples see BENSON (1976). See also transferability.
A new chemical species AB, each molecular entity of which is formed by direct combination of two separate molecular entities A and B in such a way that there is change in connectivity, but no loss, of atoms within the moieties A and B. Stoichiometries other than 1:1 are also possible, e.g. a bis-adduct (2:1). An "intramolecular adduct" can be formed when A and B are groups contained within the same molecular entity.
This is a general term which, whenever appropriate, should be used in preference to the less explicit term complex. It is also used specifically for products of an addition reaction. For examples see Lewis adduct, Meisenheimer adduct,
See energy of activation.
The term designates structures in which a hydrogen atom is bonded to both a carbon atom and a metal atom. The term is also used to characterize the interaction between a CH bond and an unsaturated metal centre, and to describe similar bonding of a transition metal with Si-H compounds. The expression "
See solvolysis.
A substitution reaction occurring at position 1/ of an allylic system, the double bond being between positions 2/ and 3/. The incoming group may be attached to the same atom 1/ as the leaving group, or the incoming group becomes attached at the relative position 3/, with movement of the double bond from 2/3 to 1/2. For example:
or
(written as a transformation).
A conjugated system of pi electrons is termed alternant if its atoms can be divided into two sets so that no atom of one set is directly linked to any other atom of the same set.
A description applied to a chemical species whose molecular entities each possess two alternative and strongly interacting distinguishable reactive centres, to either of which a bond may be made in a reaction: the centres must be connected in such a way that reaction at either site stops or greatly retards subsequent attack at the second site. The term is most commonly applied to conjugated nucleophiles, for example the enolate ion
(which may react with electrophiles either at the
Molecular entities, such as dianions of dicarboxylic acids, containing two non-interacting (or feebly interacting) reactive centres, are not generally considered to be ambident and are better described as "bifunctional".
The Latin root of the word implies two reactive centres, but the term has in the past also incorrectly been applied to chemical species with more than two reactive centres. For such species the existing term "polydent" (or, better, "multident") is more appropriate. GOMPPER (1964); SMITH and DeWALL (1977). See also chelation.
A compound containing a large organic cation or anion which possesses a long unbranched hydrocarbon chain, e.g.
The existence of distinct polar (hydrophilic) and non polar (hydrophobic) regions in the molecule promotes the formation of micelles in dilute aqueous solution.
Self-ionizing solvent possessing both characteristics of Brønsted acids and bases, for example H2O and CH3OH, in contrast to aprotic solvent.
A chemical species that behaves both as an acid and as a base is called amphoteric. This property depends upon the medium in which the species is investigated: H2SO4 is an acid when studied in water, but becomes amphoteric in superacids.
See neighbouring group participation.
anionotropic rearrangement (or anionotropy)
A rearrangement in which the migrating group moves with its electron pair from one atom to another.
See radical ion.
Alternative, but less desirable term for annulation. The term is widely used in German and French language.
A transformation involving fusion of a new ring to a molecule via two new bonds.
Some authors use the term "annelation" for the fusion of an additional ring to an already existing one, and "annulation" for the formation of a ring from one or several acyclic precursors, but this distinction is not made generally. DANHEISER, GERE and SARD (1982), JUNG (1976). See also cyclization.
Mancude (i.e. having formally the maximum number of noncumulative double bonds) monocyclic hydrocarbon without side chains of the general formula CnHn (n is an even number) or CnHn+1 (n is an odd number). Note that in systematic nomenclature an annulene with seven or more carbon atoms may be named [n]annulene, where n is the number of carbon atoms, e.g. [9]annulene for cyclonona-1,3,5,7-tetraene. IUPAC CLASS NAMES (1993). See aromatic, Hückel (4n + 2) rule.
When a part of a molecule ("molecular fragment") undergoes two changes in bonding (bond-making or bond-breaking), either to a common centre or to two related centres, external to itself, these bonding changes may be related in one of two spatially different ways. These are designated as "antarafacial" if opposite faces of the molecular fragment are involved, and "suprafacial" if both changes occur at the same face. The concept of "face" is clear from the diagrams in the cases of planar (or approximately planar) frameworks with isolated or interacting pi orbitals (Figs. a and b below).
The terms antarafacial and suprafacial are, however, also employed in cases in which the essential part of the molecular fragment undergoing changes in bonding comprises two atoms linked only by a sigma bond. In these cases it is customary to refer to the phases of the local sigma-bonding orbital: occurrence of the two bonding changes at sites of like orbital phase is regarded as suprafacial, whereas that at two sites of opposite phase is antarafacial. The possibilities are shown for C-C and C-H sigma bonds in Figs. c and d. There may be two distinct and alternative stereochemical outcomes of a suprafacial process involving a sigma bond between saturated carbon atoms, i.e. either retention or inversion at both centres. The antarafacial process results in inversion at one centre and retention at the second.
For examples of the use of these terms see cycloaddition, sigmatropic rearrangement. See also anti, sigma, pi.
In the representation of stereochemical relationships "anti" means "on opposite sides" of a reference plane, in contrast to "syn" which means "on the same side", as in the following examples.
(A) Two substituents attached to atoms joined by a single bond are anti if the torsion angle (dihedral angle) between the bonds to the substituents is greater than 90o, or syn if it is less than 90o. (A further distinction is made between antiperiplanar, synperiplanar, anticlinal and synclinal.) IUPAC ORGANIC RULES (1979); IUPAC STEREOCHEMICAL TERMINOLOGY (1993); KLYNE and PRELOG (1960).
(B) In the older literature the terms anti and syn were used to designate stereoisomers of oximes and related compounds. That usage was superseded by the terms "trans" and "cis" or E and Z, respectively.
(C) When the terms are used in the context of chemical reactions or transformations, they designate the relative orientation of substituents in the substrate or product:
(1) Addition to a carbon-carbon double bond:
(2) Alkene-forming elimination:
In the examples described under (1) and (2) anti processes are always antarafacial, and syn processes are suprafacial. See also IUPAC STEREOCHEMICAL TERMINOLOGY (1993) for further meaning of the term.
See aromatic.
See More O'Ferrall-Jencks diagram.
See Markownikoff rule.
Non-protogenic (in a given situation). (With extremely strong Brønsted acids or bases, solvents that are normally aprotic may accept or lose a proton. For example, acetonitrile is in most instances an aprotic solvent, but it is protophilic in the presence of concentrated sulfuric acid and protogenic in the presence of potassium tert-butoxide. Similar considerations apply to benzene, trichloromethane, etc.) See also dipolar aprotic solvent.
The incorporation of one or more integral molecules of water into another species with or without displacement of one or more other atoms or groups. For example the incorporation of water into the inner ligand sphere of an inorganic complex is an aquation reaction. See also hydration.
(1) In the traditional sense, "having a chemistry typified by benzene".
(2) A cyclically conjugated molecular entity with a stability (due to delocalization) significantly greater than that of a hypothetical localized structure (e.g. Kekulé structure) is said to possess aromatic character. If the structure is of higher energy (less stable) than such a hypothetical classical structure, the molecular entity is "antiaromatic".
The most widely used method for determining aromaticity is the observation of diatropicity in the 1H NMR spectrum. ATKINS (1974); GARRATT (1986). See also Hückel (4n + 2) rule, Möbius aromaticity.
(3) The terms aromatic and antiaromatic have been extended to describe the stabilization or destabilization of transition states of pericyclic reactions. The hypothetical reference structure is here less clearly defined, and use of the term is based on application of the Hückel (4n + 2) rule and on consideration of the topology of orbital overlap in the transition state. Reactions of molecules in the ground state involving antiaromatic transition states proceed, if at all, much less easily than those involving aromatic transition states. DEWAR (1971); ZIMMERMANN (1971).
See energy of activation.
A hydrocarbon derived from an arene by abstraction of two hydrogen atoms from adjacent carbon atoms; thus 1,2-didehydroarene. Arynes are commonly represented with a formal triple bond. The analogous heterocyclic compounds are called heteroarynes or hetarynes. E.g.
Arynes are usually transient species. IUPAC CLASS NAMES (1993).
The assembling of separate molecular entities into any aggregate, especially of oppositely charged free ions into ion pairs or larger and not necessarily well-defined clusters of ions held together by electrostatic attraction. The term signifies the reverse of dissociation, but is not commonly used for the formation of definite adducts by colligation or coordination.
The traditional term describing the preferential formation in a chemical reaction of one enantiomer or diastereoisomer over the other as a result of the influence of a chiral feature in the substrate, reagent, catalyst or environment. The term also refers to the formation of a new chiral feature preferentially in one configuration under such influence. IUPAC STEREOCHEMICAL TERMINOLOGY (1993).
A one-electron wavefunction describing an electron in the effective field provided by a nucleus and the other electrons present. See also molecular orbital.
A transformation by which one molecular entity (the substrate) is converted into another by the formation of one (and only one) two-centre bond between the substrate and another molecular entity and which involves no other changes in connectivity in the substrate. For example, the formation of an acyl cation by attachment of carbon monoxide to a carbenium ion (R+):
The product of an attachment may also be the adduct of the two reactants, but not all adducts can be represented as the products of an attachment. (For example, the Diels-Alder cycloaddition
results in an adduct of buta-1,3-diene and ethene, but the reaction cannot be described as an attachment since bonds are formed between more than two centres.) See also colligation, electron attachment.
A chemical reaction in which a product (or a reaction intermediate) also functions as catalyst. In such a reaction the observed rate of reaction is often found to increase with time from its initial value. See order of reaction.
Synonymous with degenerate rearrangement.
A proton (hydron) transfer reaction between two identical molecules (usually a solvent), one acting as a Brønsted acid and the other as a Brønsted base. For example:
The product of the activities (or, more approximately, concentrations) of the species produced as the result of autoprotolysis. For solvents in which no other ionization processes are significant the term is synonymous with "ionic product". The autoprotolysis constant for water, Kw, is equal to the product of activities
The conformational preference of an equatorial compared to an axial substituent in a monosubstituted cyclohexane. This steric substituent parameter equals
See nitrene.
See nitrene.
See nitrene.
A chemical reaction resulting in a single reaction product from two or three reacting chemical species, with formation of two new chemical bonds to the same atom in one of the reactant molecular entities. The synonymous term 1/1/addition is also used. For example:
(This particular example can also be viewed as an insertion reaction.) In inorganic chemistry such
A positive deviation of an
The use of the term has been extended to include the effect of any substituent on an adjacent reactive centre, for example in the case of the "
A transformation of the general type
where the central atom Z is commonly carbon. The reverse reaction is called
ATKINS, P. W. (1974), "Quanta: a Handbook of Concepts", Clarendon Press, Oxford.
BENSON, S. W. (1976), "Thermochemical Kinetics", Second Edition, Wiley-Interscience, New York.
BROOKHART, M., and GREEN, M. L. H. (1983), J. Organomet. Chem., 250, 395-408.
COX, R. A., and YATES, K. (1983), Can. J. Chem., 61, 2225-2243.
DANHEISER, R. L., GERE, S. K., and SARD, H. (1982), J. Am. Chem. Soc., 104, 7670-7672.
DEWAR, M. J. S. (1971), Angew. Chem., Int. Ed. Engl., 10, 761-776.
GARRATT, P. J. (1986), "Aromaticity", Wiley, New York.
GOMPPER, R. (1964), Angew. Chem., Int. Ed. Engl., 3, 560-570.
GUTMANN, V. (1976), Coord. Chem. Rev., 18, 225-255.
HIRSCH, J. A. (1967), Top. in Stereochem., 1, 199-222.
HOZ, S. and BUNCEL, E. (1985), Isr. J. Chem., 26, 313-319.
*IUPAC CLASS NAMES (1993). IUPAC: Organic Chemistry Division. Glossary of Class Names of Organic Compounds and Reactive Intermediates Based on Structure. IDCNS and public review; now published in Pure Appl. Chem., 67, 1307-1375 (1995).
*IUPAC ORGANIC RULES (1979). IUPAC: Nomenclature of organic chemistry: definitive rules, 1979. Sections A, B, C, D, E, F, and H. Pergamon Press, Oxford.
*IUPAC STEREOCHEMICAL TERMINOLOGY (1993). IUPAC: Organic Chemistry Division: Basic Terminology of Stereochemistry. IDCNS and public review. Now published as Basic Terminology of Stereochemistry (IUPAC Recommendations 1996) in Pure Appl. Chem., 68, 2193-2222 (1996).
JUNG, M.E. (1976), Tetrahedron, 32, 3-31.
KLYNE, W., and PRELOG, V. (1960), Experientia, 16, 521-523.
LUO, X.-L., and CRABTREE, R. H. (1989), J. Am. Chem. Soc., 111, 2527-2535.
ROCHESTER, C. H. (1970), "Acidity Functions", Academic Press, New York.
SMITH, P. A. S., and DeWALL, G. L. (1977), J. Am. Chem. Soc., 99, 5751-5760.
ZIMMERMAN, H. E. (1971), Acc. Chem. Res., 4, 272-280.
![[pi]](../greek/PI.GIF) -adducts.
-adducts.![[double dagger]](../greek/DDAGGER.GIF) , is defined as that assembly of atoms which corresponds to an arbitrary infinitesimally small region at or near the col (saddle point) of a potential energy surface. See also transition state.-addition, 1/1/addition), at two adjacent sites (1/2/addition) or at two non-adjacent sites (1/3/- or 1/4/addition, etc.). For example
, is defined as that assembly of atoms which corresponds to an arbitrary infinitesimally small region at or near the col (saddle point) of a potential energy surface. See also transition state.-addition, 1/1/addition), at two adjacent sites (1/2/addition) or at two non-adjacent sites (1/3/- or 1/4/addition, etc.). For example(a) H+ + Br- + (CH3)2C=CH2
![]() (CH3)2CBr-CH3 (1/2/addition)
(CH3)2CBr-CH3 (1/2/addition)![]() BrCH2-CH=CH-CH2Br (1/4/addition) and BrCH2-CH(Br)-CH=CH2 (1/2/addition)-addition, cheletropic reaction, cycloaddition.-adduct.
BrCH2-CH=CH-CH2Br (1/4/addition) and BrCH2-CH(Br)-CH=CH2 (1/2/addition)-addition, cheletropic reaction, cycloaddition.-adduct.![[mu]](../greek/MU.GIF) -hydrido-bridged" is also used to describe the bridging hydrogen. BROOKHART and GREEN (1983); LUO and CRABTREE (1989).
-hydrido-bridged" is also used to describe the bridging hydrogen. BROOKHART and GREEN (1983); LUO and CRABTREE (1989).![]() CH3CH=CHCH2OAc
CH3CH=CHCH2OAc![]() CH3CH(OAc)CH=CH2
CH3CH(OAc)CH=CH2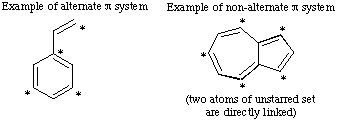
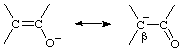
![[beta]](../greek/BETA.GIF) -carbon atom or at oxygen) or
-carbon atom or at oxygen) or ![[gamma]](../greek/GAMMA.GIF) -pyridones, and also to the vicinally ambident cyanide ion, cyanate ion, thiocyanate ion, sulfinate ion, nitrite ion, and unsymmetrical hydrazines. Ambident electrophiles are exemplified by carboxylic esters RC(O)OCR3 which react with nucleophiles either at the carbonyl carbon or the alkoxy carbon.
-pyridones, and also to the vicinally ambident cyanide ion, cyanate ion, thiocyanate ion, sulfinate ion, nitrite ion, and unsymmetrical hydrazines. Ambident electrophiles are exemplified by carboxylic esters RC(O)OCR3 which react with nucleophiles either at the carbonyl carbon or the alkoxy carbon.H3C(CH2)nCO2-M+
H3C(CH2)nSO3-M+
H3C(CH2)nN(CH3)3+X-
(n > 7).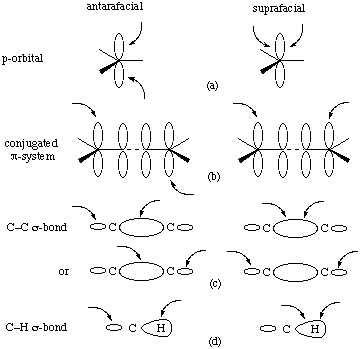
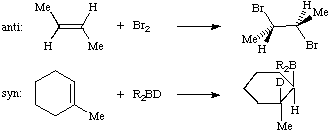
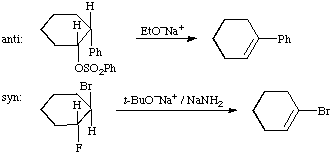
![]() (RCO)+
(RCO)+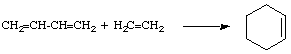
![]() H3O+ + OH-
H3O+ + OH-![[Delta]](../greek/DDELTA.GIF) rGo in kcal/mol for the equatorial to axial equilibration on cyclohexane. The values are also known as "Winstein-Holness" A values. HIRSCH (1967); CAREY and SUNDBERG (1990).
rGo in kcal/mol for the equatorial to axial equilibration on cyclohexane. The values are also known as "Winstein-Holness" A values. HIRSCH (1967); CAREY and SUNDBERG (1990).![]() Cl2CHOCH3-addition reactions, generally to a metallic central atom, are known as "oxidative additions".-Addition is the reverse of -elimination or 1/1/elimination. See also addition, elimination.-nucleophile (a nucleophile bearing an unshared pair of electrons on an atom adjacent to the nucleophilic site) from a Brønsted-type plot of lg knuc vs. pKa constructed for a series of related normal nucleophiles. More generally, it is the influence of the atom bearing a lone pair of electrons on the reactivity at the adjacent site. HOZ and BUNCEL (1985). See also Brønsted relation.-silicon effect".
Cl2CHOCH3-addition reactions, generally to a metallic central atom, are known as "oxidative additions".-Addition is the reverse of -elimination or 1/1/elimination. See also addition, elimination.-nucleophile (a nucleophile bearing an unshared pair of electrons on an atom adjacent to the nucleophilic site) from a Brønsted-type plot of lg knuc vs. pKa constructed for a series of related normal nucleophiles. More generally, it is the influence of the atom bearing a lone pair of electrons on the reactivity at the adjacent site. HOZ and BUNCEL (1985). See also Brønsted relation.-silicon effect".![]() RR'Z + XY (or X + Y, or X+ + Y-)-addition.
RR'Z + XY (or X + Y, or X+ + Y-)-addition.
References
Continue with terms starting with B.
Return to home page for Glossary of terms used in Physical Organic Chemistry.