![[sigma]](../greek/SIGMA.GIF) * delocalization (or n-* no bond resonance)
* delocalization (or n-* no bond resonance)
Contents
+ narcissistic reaction; neighbouring group participation; NHOMO; NIH shift; nitrene; nitrenium ion; no-bond resonance; nonclassical carbocation; normal kinetic isotope effect; nucleofuge; nucleophile, nucleophilic; nucleophilic catalysis; nucleophilicity; n-* delocalization (or n-* no bond resonance)
onium ion; opposing reactions; optical yield; orbital; orbital steering; orbital symmetry; order of reaction; outer-sphere (electron transfer); oxidation; oxidation number; oxidative addition; oxidative coupling
A chemical reaction that can be described as the conversion of a reactant into its mirror image, without rotation or translation of the product, so that the product enantiomer actually coincides with the mirror image of the reactant molecule. Examples of such reactions are cited under the entries fluxional and degenerate rearrangement. SALEM (1971).
neighbouring group participation
The direct interaction of the reaction centre (usually, but not necessarily, an incipient carbenium centre) with a lone pair of electrons of an atom or with the electrons of a sigma or pi bond contained within the parent molecule but not conjugated with the reaction centre. A distinction is sometimes made between n, sigma, and pi participation.
A rate increase due to neighbouring group participation is known as "anchimeric assistance". "Synartetic acceleration" is the special case of anchimeric assistance ascribed to participation by electrons binding a substituent to a carbon atom in a ![[beta]](../greek/BETA.GIF) -position relative to the leaving group attached to the
-position relative to the leaving group attached to the ![[alpha]](../greek/ALPHA.GIF) -carbon atom. According to the underlying model, these electrons then provide a three-centre bond (or "bridge") "fastening together" (as the word "synartetic" is intended to suggest) the - and -carbon atoms between which the charge is divided in the intermediate bridged ion formed (and in the transition state preceding its formation). The term synartetic acceleration is not widely used. See also intramolecular catalysis, multi-centre bond.
-carbon atom. According to the underlying model, these electrons then provide a three-centre bond (or "bridge") "fastening together" (as the word "synartetic" is intended to suggest) the - and -carbon atoms between which the charge is divided in the intermediate bridged ion formed (and in the transition state preceding its formation). The term synartetic acceleration is not widely used. See also intramolecular catalysis, multi-centre bond.
See subjacent orbital.
The intramolecular hydrogen migration which can be observed in enzymatic and chemical hydroxylations of aromatic rings. It is evidenced by appropriate deuterium labelling, i.e.
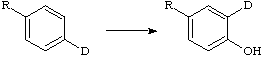
In enzymatic reactions the NIH shift is generally thought to derive from the rearrangement of arene oxide intermediates, but other pathways have been suggested.
(NIH stands for National Institutes of Health where the shift was discovered.)
HANZLIK, HOGBERG and JUDSON (1984).
Generic name for HN: and substitution derivatives thereof, containing an electrically neutral univalent nitrogen atom with four non-bonding electrons. Two of these are paired; the other two may have parallel spins (triplet state) or antiparallel spins (singlet state). The name is the strict analogue of carbene and, as a generic name, it is preferred to a number of alternatives proposed ("imene", "imine radical", "azene", "azylene", "azacarbene", "imin", "imidogen"). See LWOWSKI (1970).
The cation H2N+ and its N-hydrocarbyl derivatives R2N+, in which the nitrogen has a positive charge, and two unshared electrons. A synonymous term is aminylium ion. IUPAC CLASS NAMES (1993).
See hyperconjugation.
A carbocation the ground state of which has delocalized (bridged) bonding pi- or sigma-electrons. (N.B.: Allylic and benzylic carbocations are not considered nonclassical.) BARTLETT (1965).
See isotope effect.
A leaving group that carries away the bonding electron pair. For example, in the hydrolysis of an alkyl chloride, Cl- is the nucleofuge. The tendency of atoms or groups to depart with the bonding electron pair is called nucleofugality.
The adjective is nucleofugal. See also electrofuge, nucleophile.
A nucleophile (or nucleophilic reagent) is a reagent that forms a bond to its reaction partner (the electrophile) by donating both bonding electrons.
A "nucleophilic substitution reaction" is a heterolytic reaction in which the reagent supplying the entering group acts as a nucleophile. For example
The term "nucleophilic" is also used to designate the apparent polar character of certain radicals, as inferred from their higher relative reactivity with reaction sites of lower electron density.
Nucleophilic reagents are Lewis bases.
Catalysis by a Lewis base, involving formation of a Lewis adduct as a reaction intermediate. For example, the hydrolysis of acetic anhydride in aqueous solution catalysed by pyridine:
[C5H5NCOCH3]+ + H2O ![]() C5H5N + CH3CO2H + H+aq
C5H5N + CH3CO2H + H+aq
See also electrophilic, nucleophilicity.
(1) The property of being nucleophilic.
(2) The relative reactivity of a nucleophilic reagent. (It is also sometimes referred to as "nucleophilic power".) Qualitatively, the concept is related to Lewis basicity. However, whereas Lewis basicity is measured by relative equilibrium constants,
![[reversible arrow]](POgif/NUCL.GIF) B+-A- (equilibrium constant K)
B+-A- (equilibrium constant K)nucleophilicity of a Lewis base is measured by relative rate constants of different nucleophilic reagents towards a common substrate, most commonly involving formation of a bond to carbon,
See also electrophilicity, Ritchie equation, Swain-Scott equation.
n-* delocalization (or n-* no bond resonance)
Delocalization of a free electron pair (n) into an antibonding -orbital (s*).
See hyperconjugation, resonance.
(1) A cation (with its counterion) derived by addition of a hydron to a mononuclear parent hydride of the nitrogen, chalcogen and halogen family, e.g. H4N+ ammonium ion.
(2) Derivatives formed by substitution of the above parent ions by univalent groups, e.g. (CH3)2S+H dimethylsulfonium, (CH3CH2)4N+ tetraethylammonium.
(3) Derivatives formed by substitution of the above parent ions by groups having two or three free valencies on the same atom. Such derivatives are, whenever possible, designated by a specific class name. E.g. R2C=NH2+ iminium ion. IUPAC CLASS NAMES (1993). See also carbenium ion, carbonium ion.
See composite reactions.
In a chemical reaction involving chiral reactants and products, the ratio of the optical purity of the product to that of the precursor, reactant or catalyst. This should not be confused with "enantiomeric excess". The optical yield is in no way related to the chemical yield of the reaction. IUPAC STEREOCHEMICAL TERMINOLOGY (1993). See stereoselectivity.
See atomic orbital, molecular orbital.
A concept expressing that the stereochemistry of approach of two reacting species is governed by the most favourable overlap of their appropriate orbitals.
The behaviour of an atomic or localized molecular orbital under molecular symmetry operations characterizes its orbital symmetry. For example, under a reflection in an appropriate symmetry plane, the phase of the orbital may be unchanged (symmetric), or it may change sign (antisymmetric), i.e. the positive and negative lobes are interchanged.
A principal context for the use of orbital symmetry is the discussion of chemical changes that involve "conservation of orbital symmetry". If a certain symmetry element (e.g. the reflection plane) is retained along a reaction pathway, that pathway is "allowed" by orbital symmetry conservation if each of the occupied orbitals of the reactant(s) is of the same symmetry type as a similarly (e.g. singly or doubly) occupied orbital of the product(s). This principle permits the qualitative construction of correlation diagrams to show how molecular orbitals transform (and how their energies change) during idealized chemical changes (e.g. cycloadditions).
An idealized single bond is a sigma bond, i.e., it has cylindrical symmetry, whereas a p-orbital or pi-bond orbital has pi symmetry, i.e. it is antisymmetric with respect to reflection in a plane passing through the atomic centres with which it is associated. In ethene, the pi-bonding orbital is symmetric with respect to reflection in a plane perpendicular to and bisecting the C-C bond, whereas the pi*-antibonding orbital is antisymmetric with respect to this operation.
Considerations of orbital symmetry are frequently grossly simplified in that, for example, the pi orbitals of a carbonyl group would be treated as having the same symmetry as those of ethene, and the fact that the carbonyl group in, for example, camphor, unlike that in formaldehyde, has no mirror planes would be ignored. These simplified considerations nevertheless afford the basis of one approach to the understanding of the rules which indicate whether pericyclic reactions are likely to occur under thermal or photochemical conditions. WOODWARD and HOFFMANN (1969); HALEVI (1992). See also sigma, pi.
order of reaction, n (SI unit: 1)
If the macroscopic (observed, empirical or phenomenological) rate of reaction[B]... (expressing in full the dependence of the rate of reaction on the concentrations [A], [B]...) where , are constant exponents (independent of concentration and time) and k is independent of [A] and [B] etc. (rate constant, rate coefficient), then the reaction is said to be of order with respect to A, of order with respect to B,..., and of (total or overall) order ![[nu]](../greek/NU.GIF) = ++...., ,... can be positive or negative integral or rational nonintegral numbers. They are the reaction orders with respect to A, B,... and are sometimes called "partial orders of reaction". Orders of reaction deduced from the dependence of initial rates of reaction on concentration are called "orders of reaction with respect to concentration"; orders of reaction deduced from the dependence of the rate of reaction on time of reaction are called "orders of reaction with respect to time".
= ++...., ,... can be positive or negative integral or rational nonintegral numbers. They are the reaction orders with respect to A, B,... and are sometimes called "partial orders of reaction". Orders of reaction deduced from the dependence of initial rates of reaction on concentration are called "orders of reaction with respect to concentration"; orders of reaction deduced from the dependence of the rate of reaction on time of reaction are called "orders of reaction with respect to time".
The concept of order of reaction is also applicable to chemical rate processes occurring in systems for which concentration changes (and hence the rate of reaction) are not themselves measurable, provided it is possible to measure a chemical flux. For example, if there is a dynamic equilibrium according to the equation
![[reversible arrow]](../greek/REVARR.GIF) pP
pPand if a chemical flux is experimentally found, (e.g. by NMR line shape analysis) to be related to concentrations by the equation
![[phi]](../greek/PHI.GIF) -A/a = k[A][L]
-A/a = k[A][L]![[lambda]](../greek/LAMBDA.GIF) ,
,
then the corresponding reaction is of order with respect to A... and of total (or overall) order (= + +...)
The proportionality factor k above is called the (nth order) "rate coefficient".
Rate coefficients referring to (or believed to refer to) elementary reactions are called "rate constants" or, more appropriately "microscopic" (hypothetical, mechanistic) rate constants.
The (overall) order of a reaction cannot be deduced from measurements of a "rate of appearance" or "rate of disappearance" at a single value of the concentration of a species whose concentration is constant (or effectively constant) during the course of the reaction. If the overall rate of reaction is, for example, given by
[B]
but [B] stays constant, then the order of the reaction (with respect to time), as observed from the concentration change of A with time, will be , and the rate of disappearance of A can be expressed in the form
The proportionality factor kobs deduced from such an experiment is called the "observed rate coefficient" and it is related to the ( + )th order rate coefficient k by the equation
For the common case when = 1, kobs is often referred to as a "pseudo-first order rate coefficient" (k![[psi]](../greek/PSI.GIF) ).
).
For a simple (elementary) reactions a partial order of reaction is the same as the stoichiometric number of the reactant concerned and must therefore be a positive integer (see rate of reaction). The overall order is then the same as the molecularity. For stepwise reactions there is no general connection between stoichiometric numbers and partial orders. Such reactions may have more complex rate laws, so that an apparent order of reaction may vary with the concentrations of the chemical species involved and with the progress of the reaction: in such cases it is not useful to speak of orders of reaction, although apparent orders of reaction may be deducible from initial rates.
In a stepwise reaction, orders of reaction may in principle always be assigned to the elementary steps. See also kinetic equivalence.
outer-sphere (electron transfer)
An outer-sphere electron transfer is a reaction in which the electron transfer takes place with no or very weak (4 -16 kJ mol-1) electronic interaction between the reactants in the transition state. If instead the donor and the acceptor exhibit a strong electronic coupling, the reaction is described as inner-sphere electron transfer. The two terms derive from studies concerning metal complexes and it has been suggested that for organic reactions the term "nonbonded" and "bonded" electron transfer should be used. See also inner-sphere electron transfer. EBERSON (1987); LITTLER (1970).
(1) The complete, net removal of one or more electrons from a molecular entity (also called "de-electronation").
(2) an increase in the oxidation number of any atom within any substrate (see HENDRICKSON, CRAM and HAMMOND (1970)).
(3) Gain of oxygen and/or loss of hydrogen of an organic substrate.
All oxidations meet criteria (1) and (2), and many meet criterion (3), but this is not always easy to demonstrate.Alternatively, an oxidation can be described as a transformation of an organic substrate that can be rationally dissected into steps or primitive changes. The latter consist in removal of one or several electrons from the substrate followed or preceded by gain or loss of water and/or hydrons or hydroxide ions, or by nucleophilic substitution by water or its reverse and/or by an intramolecular molecular rearrangement.
This formal definition allows the original idea of oxidation (combination with oxygen), together with its extension to removal of hydrogen, as well as processes closely akin to this type of transformation (and generally regarded in current usage of the term in organic chemistry to be oxidations and to be effected by "oxidizing agents") to be descriptively related to definition (1). For example the oxidation of methane to chloromethane may be considered as follows:
See IUPAC INORGANIC RULES (1970), Rule 0.1; IUPAC INORGANIC NOMENCLATURE (1990), Rules I-5.5.2 and I-10.2.7. See also oxidation.
The insertion of a metal of a metal complex into a covalent bond involving formally an overall two-electron loss on one metal or a one-electron loss on each of two metals, i.e.,
2 LnMm + XY ![]() LnMm+1(X) + LnMm+1(Y)
LnMm+1(X) + LnMm+1(Y)
In free-radical chemistry, the term is used to indicate a free radical addition to a carbon-carbon double bond, under oxidative conditions. For example:
The coupling of two molecular entities through an oxidative process, usually catalysed by a transition metal compound and involving dioxygen as the oxidant; e.g.,
BARTLETT, P. D. (Ed.) (1965), "Nonclassical Ions", Benjamin, New York.
EBERSON, L. (1987), "Electron Transfer Reactions in Organic Chemistry", Springer, Berlin.
HALEVI, E. A. (1992), "Orbital Symmetry and Reaction Mechanisms. The Ocams View", Springer, Berlin.
HANZLIK, R. P., HOGBERG, K., and JUDSON, C. M. (1984), Biochemistry, 23, 3048-3055.
*IUPAC CLASS NAMES (1993). IUPAC: Organic Chemistry Division. Glossary of Class Names of Organic Compounds and Reactive Intermediates Based on Structure. IDCNS and public review; now published in Pure Appl. Chem., 67, 1307-1375 (1995).
*IUPAC STEREOCHEMICAL TERMINOLOGY (1993). IUPAC: Organic Chemistry Division: Basic Terminology of Stereochemistry. IDCNS and public review. Now published as Basic Terminology of Stereochemistry (IUPAC Recommendations 1996) in Pure Appl. Chem., 68, 2193-2222 (1996).
LWOWSKI, W. (1970), "Nitrenes", Interscience, New York.
SALEM, L. (1971), Acc. Chem. Res., 4, 322-328.
WOODWARD, R. B., and HOFFMANN, R. (1969), Angew. Chem., Int. Ed. Engl., 8, 781-853.
Return to home page for Glossary of terms used in Physical Organic Chemistry.