GLOSSARY OF TERMS USED IN PHYSICAL ORGANIC CHEMISTRY
(IUPAC Recommendations 1994)
Sa to Sp
Continued from terms starting with R
Contents
sacrificial hyperconjugation; + salt effect; saturation transfer; Saytzeff rule; scavenger; scrambling; secondary kinetic electrolyte effect; secondary kinetic isotope effect; selectivity; selectivity factor; shielding; sigma, pi (![[sigma]](../greek/SIGMA.GIF) ,
, ![[pi]](../greek/PI.GIF) ); sigmatropic rearrangement; silylene; single-electron transfer mechanism (SET); single-step reaction; Slater-type orbital; soft acid; soft base; solvation; solvatochromic relationship; solvatochromism; solvent isotope effect; solvent parameter; solvent polarity; solvent-separated ion pair; solvolysis; solvophobicity parameter; SOMO; sonication; special salt effect; species; specific catalysis; spectator mechanism; spin adduct; spin counting; spin density; spin label; spin trapping
); sigmatropic rearrangement; silylene; single-electron transfer mechanism (SET); single-step reaction; Slater-type orbital; soft acid; soft base; solvation; solvatochromic relationship; solvatochromism; solvent isotope effect; solvent parameter; solvent polarity; solvent-separated ion pair; solvolysis; solvophobicity parameter; SOMO; sonication; special salt effect; species; specific catalysis; spectator mechanism; spin adduct; spin counting; spin density; spin label; spin trapping
sacrificial hyperconjugation
See hyperconjugation.
+ salt effect
See kinetic electrolyte effect.
saturation transfer
A term used in nuclear magnetic resonance. When a nucleus is strongly irradiated, its spin population may partly be transferred to another nucleus by an exchange process. See magnetization transfer.
Saytzeff rule
Dehydrohalogenation of secondary- and tertiary-alkyl halides proceeds by the preferential removal of the ![[beta]](../greek/BETA.GIF) -hydrogen from the carbon that has the smallest number of hydrogens. Originally formulated by A. Saytzeff (Zaitsev) to generalize the orientation in -elimination reactions of alkyl halides, this rule has been extended and modified, as follows: When two or more olefins can be produced in an elimination reaction, the thermodynamically most stable alkene will predominate. Exceptions to the Saytzeff rule are exemplified by the Hofmann rule. SAYTZEFF (1875). See also Markownikoff rule.
-hydrogen from the carbon that has the smallest number of hydrogens. Originally formulated by A. Saytzeff (Zaitsev) to generalize the orientation in -elimination reactions of alkyl halides, this rule has been extended and modified, as follows: When two or more olefins can be produced in an elimination reaction, the thermodynamically most stable alkene will predominate. Exceptions to the Saytzeff rule are exemplified by the Hofmann rule. SAYTZEFF (1875). See also Markownikoff rule.
scavenger
A substance that reacts with (or otherwise removes) a trace component (as in the scavenging of trace metal ions) or traps a reactive reaction intermediate. IUPAC COMPENDIUM (1987). See also inhibition.
scrambling
See isotopic scrambling.
secondary kinetic electrolyte effect
See kinetic electrolyte effect.
secondary kinetic isotope effect
See isotope effect.
selectivity
The discrimination shown by a reagent in competitive attack on two or more substrates or on two or more positions in the same substrate. It is quantitatively expressed by ratios of rate constants of the competing reactions, or by the decadic logarithms of such ratios.
See also isoselective relationship, partial rate factor, regioselectivity, selectivity factor, stereoselectivity.
selectivity factor
A quantitative representation of selectivity in aromatic substitution reactions (usually electrophilic, for monosubstituted benzene derivatives). If the partial rate factor, f, expresses the reactivity of a specified position in the aromatic compound PhX relative to that of a single position in benzene, then the selectivity factor Sf (expressing discrimination between p- and m-positions in PhX) is defined as
Sf = lg (fpX/fmX).
STOCK and BROWN (1963).
shielding
In the context of NMR spectroscopy shielding is the effect of the electron shells of the observed and the neighbouring nuclei on the external magnetic field. The external field induces circulations in the electron cloud. The resulting magnetic moment is oriented in the opposite direction to the external field, so that the local field at the central nucleus is weakened, although it may be strengthened at other nuclei (deshielding).
The phenomenon is the origin of the structural dependence of the resonance frequencies of the nuclei. See also chemical shift.
sigma, pi (, )
The terms are symmetry designations, pi molecular orbitals being antisymmetric with respect to a defining plane containing at least one atom (e.g. the molecular plane of ethene), and sigma molecular orbitals symmetric with respect to the same plane. In practice the terms are used both in this rigorous sense (for orbitals encompassing the entire molecule) and also for localized two-centre orbitals or bonds, and it is necessary to make a clear distinction between the two usages.
In the case of two-centre bonds, a pi bond has a nodal plane that includes the internuclear bond axis, whereas a sigma bond has no such nodal plane. (A delta bond in organometallic or inorganic molecular species has two nodes.) Radicals are classified by analogy into sigma and pi radicals.
Such two-centre orbitals may take part in molecular orbitals of sigma or pi symmetry. For example, the methyl group in propene contains three C-H bonds, each of which is of local sigma symmetry (i.e. without a nodal plane including the internuclear axis), but these three "sigma bonds" can in turn be combined to form a set of group orbitals one of which has pi symmetry with respect to the principal molecular plane and can accordingly interact with the two-centre orbital of pi symmetry (pi bond) of the double-bonded carbon atoms, to form a molecular orbital of pi symmetry.
Such an interaction between the CH3 group and the double bond is an example of what is called hyperconjugation. This cannot rigorously be described as "sigma-pi conjugation" since sigma and pi here refer to different defining planes, and interaction between orbitals of different symmetries (with respect to the same defining plane) is forbidden. See also JORGENSEN and SALEM (1973).
sigmatropic rearrangement
A molecular rearrangement that involves both the creation of a new sigma bond between atoms previously not directly linked and the breaking of an existing sigma bond. There is normally a concurrent relocation of pi bonds in the molecule concerned, but the total number of pi and sigma bonds does not change. The term was originally restricted to intramolecular pericyclic reactions, and many authors use it with this connotation. It is, however, also applied in a more general, purely structural, sense.
If such reactions are intramolecular, their transition state may be visualized as an association of two fragments connected at their termini by two partial sigma bonds, one being broken and the other being formed as, for example, the two allyl fragments in (a'). Considering only atoms within the (real or hypothetical) cyclic array undergoing reorganization, if the numbers of these in the two fragments are designated i and j, then the rearrangement is said to be a sigmatropic change of order [i,j] (conventionally [i] <= [j]). Thus the rearrangement (a) is of order [3,3], whilst reaction (b) is a [1,5]sigmatropic shift of hydrogen. (N.B.: By convention square brackets [...] here refer to numbers of atoms, in contrast with current usage in the context of cycloaddition.)
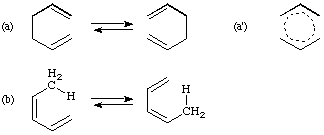
The descriptors a and s (antarafacial and suprafacial) may also be annexed to the numbers i and j; (b) is then described as a [1s,5s] sigmatropic rearrangement, since it is suprafacial with respect both to the hydrogen atom and to the pentadienyl system:
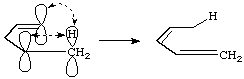
The prefix "homo" (meaning one extra atom, interrupting conjugation - cf. "homoaromaticity") has frequently been applied to sigmatropic rearrangements, but is misleading. See also cycloaddition, tautomerism.
silylene
(1) Generic name for H2Si: and substitution derivatives thereof, containing an electrically neutral bivalent silicon atom with two non-bonding electrons. (The definition is analogous to that given for carbene.)
(2) The silanediyl group (H2Si), analogous to the methylene group (H2C).
single-electron transfer mechanism (SET)
A reaction mechanism characterized by the transfer of a single electron between the species occurring on the reaction coordinate of one of the elementary steps.
single-step reaction
A reaction that proceeds through a single transition state.
Slater-type orbital
An approximate atomic orbital that attempts to allow for electron-electron repulsion by scaling the nuclear charge for each orbital.
soft acid
See hard acid.
soft base
See hard base.
solvation
Any stabilizing interaction of a solute (or solute moiety) and the solvent or a similar interaction of solvent with groups of an insoluble material (i.e., the ionic groups of an ion-exchange resin). Such interactions generally involve electrostatic forces and van der Waals forces, as well as chemically more specific effects such as hydrogen bond formation. See also cybotactic region.
solvatochromic relationship
A linear free-energy relationship based on solvatochromism. See also Kamlet-Taft solvent parameters.
solvatochromism
The (pronounced) change in position and sometimes intensity of an electronic absorption or emission band, accompanying a change in the polarity of the medium. Negative (positive) solvatochromism corresponds to a hypsochromic (bathochromic) shift with increasing solvent polarity. BUNCEL and RAJAGOPAL (1990); REICHARDT (1988). See also Dimroth-Reichardt ET parameter, Z-value.
solvent isotope effect
See isotope effect.
solvent parameter
Quantitative measures of the capability of solvents for interaction with solutes. Such parameters have been based on numerous different physico-chemical quantities, e.g. rate constants, solvatochromic shifts in ultraviolet/visible spectra, solvent-induced shifts in infrared frequencies, etc. Some solvent parameters are purely empirical in nature, i.e. they are based directly on some experimental measurement. It may be possible to interpret such a parameter as measuring some particular aspect of solvent-solute interaction or it may be regarded simply as a measure of solvent polarity. Other solvent parameters are based on analysing experimental results. Such a parameter is considered to quantify some particular aspect of solvent capability for interaction with solutes. See REICHARDT (1965). See also Dimroth-Reichardt ET parameter, Grunwald-Winstein equation, Kamlet-Taft solvent parameters, Koppel-Palm solvent parameters, solvophobicity parameter, Z-value.
solvent polarity
See polarity.
solvent-separated ion pair
See ion pair.
solvolysis
Generally, reaction with a solvent, or with a lyonium ion or lyate ion, involving the rupture of one or more bonds in the reacting solute. More specifically the term is used for substitution, elimination and fragmentation reactions in which a solvent species is the nucleophile ("alcoholysis" if the solvent is an alcohol, etc.).
solvophobicity parameter
A solvent parameter defined by
Sp = 1 - M/M(hexadecane)
derived from the Gibbs energy of transfer (![[Delta]](../greek/DDELTA.GIF) tGo) of a series of solutes from water to numerous aqueous-organic mixtures and to pure solvents:
tGo) of a series of solutes from water to numerous aqueous-organic mixtures and to pure solvents:
tGo(to solvent) = MRT + D
where RT is a solute parameter, and M and D characterize the solvent. The M values are used to define a solvent solvophobic effect so that Sp values are scaled from unity (water) to zero (hexadecane). ABRAHAM, GRELLIER, and MCGILL (1988).
SOMO
A Singly Occupied Molecular Orbital (such as the half-filled HOMO of a radical). See also frontier orbitals.
sonication
Irradiation with (often ultra)sound waves, e.g. to increase the rate of a reaction or to prepare vesicles in mixtures of surfactants and water.
special salt effect
The initial steep rate increase observed in the kinetic electrolyte effect on certain solvolysis reactions, upon addition of some non-common ion salts, especially LiClO4.
species
See chemical species.
specific catalysis
The acceleration of a reaction by a unique catalyst, rather than by a family of related substances. The term is most commonly used in connection with specific hydrogen-ion or hydroxide-ion (lyonium ion or lyate ion) catalysis. See also general acid catalysis, general base catalysis, pseudo-catalysis.
spectator mechanism
A pre-association mechanism in which one of the molecular entities, C, is already present in an encounter pair with A during formation of B from A, but does not assist the formation of B, e.g.
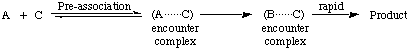
The formation of B from A may itself be a bimolecular reaction with some other reagent. Since C does not assist the formation of A, it is described as being present as a spectator, and hence such a mechanism is sometimes referred to as a spectator mechanism. See also microscopic diffusion control .
spin adduct
See spin trapping.
spin counting
See spin trapping.
spin density
The unpaired electron density at a position of interest, usually at carbon, in a radical. It is often measured experimentally by electron paramagnetic resonance (EPR, ESR (electron spin resonance)) spectroscopy through hyperfine coupling constants of the atom or an attached hydrogen. See also radical centre.
spin label
A stable paramagnetic group (typically a nitryl radical) that is attached to a part of a molecular entity whose microscopic environment is of interest and may be revealed by the electron spin resonance (ESR) spectrum of the spin label.
When a simple paramagnetic molecular entity is used in this way without covalent attachment to the molecular entity of interest it is frequently referred to as a "spin probe".
spin trapping
In certain reactions in solution a transient radical will interact with a diamagnetic reagent to form a more persistent radical. The product radical accumulates to a concentration where detection and, frequently, identification are possible by EPR/ESR spectroscopy. The key reaction is usually one of attachment; the diamagnetic reagent is said to be a "spin trap", and the persistent product radical is then the "spin adduct". The procedure is referred to as spin trapping, and is used for monitoring reactions involving the intermediacy of reactive radicals at concentrations too low for direct observation. Typical spin traps are C-nitroso compounds and nitrones, to which reactive radicals will rapidly add to form nitryl radicals. A quantitative development, in which essentially all reactive radicals generated in a particular system are intercepted, has been referred to as "spin counting". Spin trapping has also been adapted to the interception of radicals generated in both gaseous and solid phases. In these cases the spin adduct is in practice transferred to a liquid solution for observation in order to facilitate interpretation of the EPR/ESR spectra of the radicals obtained.
References
ABRAHAM, M. H., GRELLIER, P. L., and McGILL, R. A. (1988), J. Chem. Soc., Perkin Trans. II, 339-345.
BUNCEL, E., and RAJAGOPAL, S. (1990), Acc. Chem. Res., 23, 226-231.
*IUPAC COMPENDIUM (1987). IUPAC: Compendium of Chemical Terminology, (GOLD, V., LOENING, K. L., MCNAUGHT, A. D., and SEHMI, P., Eds.), Blackwell, Oxford.
JORGENSEN, W. L., and SALEM, L. (1973), "The Organic Chemist's Book of Orbitals", Academic Press, New York.
REICHARDT, C. (1965), Angew. Chem., Int. Ed. Engl., 4, 29-40.
REICHARDT, C. (1988), "Solvents and Solvent Effects in Organic Chemistry" 2nd ed., VCH Verlagsgesellschaft, Weinheim, Chap. 7.
SAYTZEFF, A. (1875), Justus Liebigs Ann. Chem., 179, 296-301.
STOCK, L. M., and BROWN, H. C. (1963), Adv. Phys. Org. Chem., 1, 35-154.
Continue with terms starting with St to Sy and .
Return to home page for Glossary of terms used in Physical Organic Chemistry.