![[sigma]](../greek/SIGMA.GIF) * and the steric substituent constant Es
* and the steric substituent constant Es
Contents
T-jump; Taft equation; + tautomeric effect; tautomerism; tautomerization; tele-substitution; telomerization; termination; tetrahedral intermediate; thermodynamic control (of product composition); thermolysis; tight-ion pairT-jump; ; torquoselectivity; transferability; transformation; transient (chemical) species; transition coordinate; transition state; transition state analogue; transition structure; transmission coefficient; transport control; trapping; tunnelling; umpolung; unimolecular; unreactive; unstable; upfield; valence; valence isomer; valence tautomerization; van der Waals forces; volume of activation; Wheland intermediate; Woodward-Hoffmann rules; Ylide; Yukawa-Tsuno equation; Z-value; Zaitsev rule; zwitterionic compound; Zucker-Hammett hypothesis; Z-value
See chemical relaxation.
Various equations are associated with R.W. Taft, but the term is most often used to designate the family of equations that emerged from Taft's analysis of the reactivities of aliphatic esters, and which involved the polar substituent constant * and the steric substituent constant Es
![[rho]](../greek/RHO.GIF) ** +
** + ![[delta]](../greek/DELTA.GIF) Es
Es
or the one-parameter forms applicable when the role of either the polar term or the steric term may be neglected. Nowadays * is usually replaced by the related constant I. TAFT (1952, 1953). See also Hammett equation, -value, -constant.
See electromeric effect.
Isomerism of the general form
![[reversible arrow]](../greek/REVARR.GIF) X=Y-Z-G
X=Y-Z-Gwhere the isomers (called tautomers) are readily interconvertible; the atoms connecting the groups X,Y,Z are typically any of C, H, O, or S, and G is a group which becomes an electrofuge or nucleofuge during isomerization. The commonest case, when the electrofuge is H+, is also known as "prototropy".
Examples, written so as to illustrate the general pattern given above, include:
Keto-enol tautomerism, such as
(CH3)C(=O)-CH2-CO2Et (keto)(G = H, X = O, Y = CCH3, Z = CHCO2Et)
ArCH2-N=CHAr' ArCH=N-CH2Ar'
(G = H, X = CHAr, Y = N, Z = CHAr')
The grouping Y may itself be a three-atom (or five-atom) chain extending the conjugation, as in
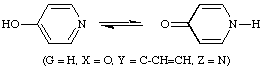
The double bond between Y and Z may be replaced by a ring, when the phenomenon is called ring-chain tautomerism, as in
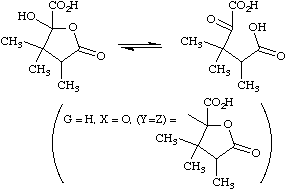
INGOLD (1953). See also ambident, sigmatropic rearrangement, tautomerization, valence tautomerization.
The isomerization by which tautomers are interconverted. It is a heterolytic molecular re-arrangement and is frequently very rapid. See tautomerism.
A substitution reaction in which the entering group takes up a position more than one atom away from the atom to which the leaving group was attached.
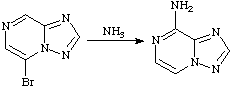
See also cine-substitution.
The formation of an addition oligomer, having uniform end groups X'...X", by a chain reaction in which a chain transfer limits the length of the polymer ("telomer") produced. An example is the polymerization of styrene in bromotrichloromethane solution (X' = CCl3, X" = Br), where Cl3C. radicals are formed in the initiation step to produce Cl3C[CH2CHPh]nBr, with n greater than 1 and often less than ca. 10:
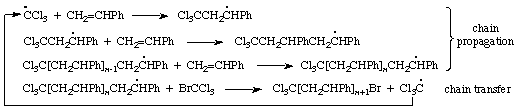
The steps in a chain reaction in which reactive intermediates are destroyed or rendered inactive, thus ending the chain.
A reaction intermediate in which the bond arrangement around an initially double-bonded carbon atom (typically a carbonyl carbon atom) has been transformed from trigonal to tetrahedral. For example, aldol in the condensation reaction of acetaldehyde (but most tetrahedral intermediates have a more fleeting existence).
thermodynamic control (of product composition)
The term characterizes conditions that lead to reaction products in a proportion governed by the equilibrium constant for their interconversion and/or for the interconversion of reaction intermediates formed in or after the rate-limiting step. (Some workers prefer to describe this phenomenon as "equilibrium control".)
See also kinetic control.
The uncatalysed cleavage of one or more covalent bonds resulting from exposure of a compound to a raised temperature, or a process in which such cleavage is an essential part.
See also pyrolysis.
See ion pair.
See chemical relaxation.
The term refers to the preference for "inward" or "outward" rotation of substituents in conrotatory or disrotatory electrocyclic ring opening reactions. JEFFORD, BERNARDINELLI, WANG, SPELLMEYER, BUDA, and HOUK (1992).
Transferability assumes invariance of properties, associated conceptually with an atom or a fragment present in a variety of molecules. The property, such as electronegativity, nucleophilicity, NMR chemical shift, etc. is held as retaining a similar value in all these occurrences.
The conversion of a substrate into a particular product, irrespective of reagents or mechanisms involved. For example, the transformation of aniline (C6H5NH2) into N-phenylacetamide (C6H5NHCOCH3) may be effected by use of acetyl chloride or acetic anhydride or ketene. A transformation is distinct from a reaction, the full description of which would state or imply all the reactants and all the products. For nomenclature of transformations see IUPAC TERMINOLOGY FOR TRANSFORMATIONS (1989).
Relating to a short-lived reaction intermediate. It can be defined only in relation to a time scale fixed by the experimental conditions and the limitations of the technique employed in the detection of the intermediate. The term is a relative one.
Transient species are sometimes also said to be "metastable". However, this latter term should be avoided, because it relates a thermodynamic term to a kinetic property, although most transients are also thermodynamically unstable with respect to reactants and products. See also persistent.
The reaction coordinate at the transition state corresponding to a vibration with an imaginary frequency. Motion along it in the two opposite senses leads towards the reactants or towards the products. See also reaction coordinate, transition state.
In theories describing elementary reactions it is usually assumed that there is a transition state of more positive molar Gibbs energy between the reactants and the products through which an assembly of atoms (initially composing the molecular entities of the reactants) must pass on going from reactants to products in either direction. In the formalism of "transition state theory" the transition state of an elementary reaction is that set of states (each characterized by its own geometry and energy) which an assembly of atoms, when randomly placed there, would have an equal probability of forming the reactants or of forming the products of that elementary reaction. The transition state is characterized by one and only one imaginary frequency. The assembly of atoms at the transition state has been called an activated complex. (It is not a complex according to the definition in this Glossary.)
It may be noted that the calculations of reaction rates by the transition state method and based on calculated potential-energy surfaces refer to the potential energy maximum at the saddle point, as this is the only point for which the requisite separability of transition state coordinates may be assumed. The ratio of the number of assemblies of atoms that pass through to the products to the number of those that reach the saddle point from the reactants can be less than unity, and this fraction is the "transmission coefficient" ![[kappa]](../greek/KAPPA.GIF) . (There are also reactions, such as the gas-phase colligation of simple radicals, that do not require "activation" and which therefore do not involve a transition state.) See also Gibbs energy of activation, Hammond principle, potential energy profile, transition structure.
. (There are also reactions, such as the gas-phase colligation of simple radicals, that do not require "activation" and which therefore do not involve a transition state.) See also Gibbs energy of activation, Hammond principle, potential energy profile, transition structure.
A substrate designed to mimic the properties or the geometry of the transition state of reaction.
A saddle point on a potential-energy surface. It has one negative force constant in the harmonic force constant matrix. See also activated complex, transition state.
See transition state.
See microscopic diffusion control.
The interception of a reactive molecule or reaction intermediate so that it is removed from the system or converted into a more stable form for study or identification. See also scavenger.
The process by which a particle or a set of particles crosses a barrier on its potential energy surface without having the energy required to surmount this barrier. Since the rate of tunnelling decreases with increasing reduced mass, it is significant in the context of isotope effects of hydrogen isotopes.
Any process by which the normal alternating donor and acceptor reactivity pattern of a chain, which is due to the presence of O or N heteroatoms, is interchanged. Reactivity umpolung is most often achieved by temporary exchange of heteroatoms (N, O) by others, such as P, S, and Se.
The original meaning of the term has been extended to the reversal of any commonly accepted reactivity pattern. For example, reaction of R-C![[triple bond]](../greek/TRIPLEB.GIF) CX (X = halide) as a synthon for "R-CC+ (i.e. electrophilic acetylene) is an umpolung of the normal more common acetylide, R-CC- (i.e. nucleophilic) reactivity. SEEBACH (1979).
CX (X = halide) as a synthon for "R-CC+ (i.e. electrophilic acetylene) is an umpolung of the normal more common acetylide, R-CC- (i.e. nucleophilic) reactivity. SEEBACH (1979).
See molecularity.
Failing to react with a specified chemical species under specified conditions. The term should not be used in place of stable, since a relatively more stable species may nevertheless be more reactive than some reference species towards a given reaction partner.
The opposite of stable, i.e. the chemical species concerned has a higher molar Gibbs energy than some assumed standard. The term should not be used in place of reactive or transient, although more reactive or transient species are frequently also more unstable.
(Very unstable chemical species tend to undergo exothermic unimolecular decompositions. Variations in the structure of the related chemical species of this kind generally affect the energy of the transition states for these decompositions less than they affect the stability of the decomposing chemical species. Low stability may therefore parallel a relatively high rate of unimolecular decomposition.)
See chemical shift.
The maximum number of univalent atoms (originally hydrogen or chlorine atoms) that may combine with an atom of the element under consideration, or with a fragment, or for which an atom of this element can be substituted.
A constitutional isomer interrelated with another by pericyclic reactions. For example, Dewar benzene, prismane and benzvalene are valence isomers of benzene.
See tautomerism.
The term describes simple reversible and generally rapid isomerizations or degenerate re-arrangements involving the formation and rupture of single and/or double bonds, without migration of atoms or groups; e.g.
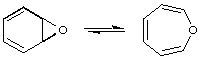
TISLER (1973) See also fluxional, tautomerism.
The attractive or repulsive forces between molecular entities (or between groups within the same molecular entity) other than those due to bond formation or to the electrostatic interaction of ions or of ionic groups with one another or with neutral molecules. The term includes: dipole-dipole, dipole-induced dipole and London (instantaneous induced dipole-induced dipole) forces.
The term is sometimes used loosely for the totality of nonspecific attractive or repulsive intermolecular forces.
A quantity derived from the pressure dependence of the rate constant of a reaction (mainly used for reactions in solution), defined by the equation
![[Delta]](../greek/DDELTA.GIF)
![[double dagger]](../greek/DDAGGER.GIF) V = -RT(
V = -RT(![[partial differential]](../greek/PARDIF.GIF) ln k/p)T
ln k/p)Tproviding that the rate constants of all reactions (except first-order reactions) are expressed in pressure-independent concentration units, such as mol dm- 3 at a fixed temperature and pressure.
The volume of activation is interpreted, according to transition state theory as the difference between the partial molar volumes of the transition state (V) and the sums of the partial volumes of the reactants at the same temperature and pressure, i.e.,
V = V - ![[Sigma]](../greek/SSIGMA.GIF) (rVR)
(rVR)where r is the order in the reactant R and VR its partial molar volume.
See Meisenheimer adduct, -adduct.
See orbital symmetry.
A chemical species produced (actually or notationally) by loss of a hydron from an atom directly attached to the central atom of an "onium ion, e.g.
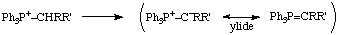
A multiparameter extension of the Hammett equation to quantify the role of enhanced resonance effects on the reactivity of meta- and para-substituted benzene derivatives, e.g.
+ r (+ - )]
The parameter r gives the enhanced resonance effect on the scale (+-), or (--), respectively.
See also -value, -constant. See YUKAWA and TSUNO (1959); SHORTER (1973).
Z-value
See below under Z-value.
See Saytzeff rule.
A neutral compound having electrical charges of opposite sign, delocalized or not on adjacent or nonadjacent atoms. Zwitterionic compounds have no uncharged canonical representations. Sometimes referred to as inner salts, ampholytes, dipolar ions (a misnomer).
For example: H3N+CH2C(=O)O-, glycine.
See also ylide.
This hypothesis states that, if in an acid catalyzed reaction, lg k1 (first-order rate constant of the reaction) is linear in Ho (Hammett acidity function), water is not involved in the transition state of the rate-controlling step. However, if lg k1 is linear in lg[H+], then water is involved. This has been shown to be incorrect by Hammett himself (see HAMMETT (1970)). LONG and PAUL (1957).
Z-value
An index of the ionizing power of a solvent based on the frequency of the longest wavelength electronic absorption maximum of 1-ethyl-4-methoxycarbonylpyridinium iodide in the solvent. The Z-value is defined by
![[lambda]](../greek/LAMBDA.GIF)
where Z is in kcal mol-1 and is in nm.
KOSOWER (1958). See also Dimroth-Reichardt ET parameter, Grunwald-Winstein equation.
KOSOWER, E. M. (1958), J. Am. Chem. Soc., 80, 3253-3260.
LONG, F. A., and PAUL, M. A. (1957), Chem. Rev., 57, 935-1010.
SEEBACH, D. (1979), Angew. Chem., Int. Ed. Engl., 18, 239-258.
TAFT, R. W., Jr. (1952, 1953), J. Am. Chem. Soc., 74, 3120-3128; 75, 4231-4238.
TISLER, M. (1973), Synthesis, 123-136.
YUKAWA. Y., and TSUNO, Y. (1959), Bull. Chem. Soc. Jpn, 32, 971-981.
Return to home page for Glossary of terms used in Physical Organic Chemistry.