![[pi]](../greek/PI.GIF) -adduct (pi-adduct); -bond (pi bond); + -complex; -electron acceptor, [pi]-electron donor group; -orbital; quantitative structure-activity relationships (QSAR); quantum yield
-adduct (pi-adduct); -bond (pi bond); + -complex; -electron acceptor, [pi]-electron donor group; -orbital; quantitative structure-activity relationships (QSAR); quantum yield
Contents
parallel reaction; paramagnetic; partial rate factor; pericyclic reaction; periselectivity; perpendicular effect; persistent; pH-rate profile; phase-transfer catalysis; phenonium ion; photolysis; + polar aprotic solvent; polar effect; polar solvent; polarity; polarizability; polydent; potential-energy profile; potential-energy (reaction) surface; pre-association; precursor complex; pre-equilibrium (or prior equilibrium); pre-exponential factor; primary kinetic electrolyte effect; primary kinetic isotope effect; primitive change; prior equilibrium; product-determining step; product development control; promotion; propagation; protic; protogenic (solvent); + protolysis; proton affinity; proton transfer reaction; protophilic (solvent); prototropic rearrangement (or prototropy); pseudo-catalysis; pseudo-first order rate coefficient; + pseudomolecular rearrangement; pseudopericyclic; + pseudo-unimolecular; pyrolysis; -adduct (pi-adduct); -bond (pi bond); + -complex; -electron acceptor, [pi]-electron donor group; -orbital; quantitative structure-activity relationships (QSAR); quantum yield
See composite reaction.
Substances having a magnetic susceptibility greater than 0 are paramagnetic. They are drawn into a magnetic field. See also diamagnetic.
The rate of substitution at one specific site in an aromatic compound relative to the rate of substitution at one position in benzene. For example, the partial rate factor fpZ for para-substitution in a monosubstituted benzene C6H5Z is related to the rate constants k(C6H5Z) and k(C6H6) for the total reaction (i.e. at all positions) of C6H5Z and benzene, respectively, and% para (the percentage para-substitution in the total product formed from C6H5Z) by the relation
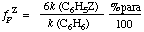
Similarly for meta-substitution:
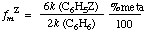
(The symbols pfZ, mfZ, ofZ are also in use.) The term applies equally to the ipso position, and it can be extended to other substituted substrates undergoing parallel reactions at different sites with the same reagent according to the same rate law. INGOLD (1953); STOCK and BROWN (1963). See also selectivity.
A chemical reaction in which concerted reorganization of bonding takes place throughout a cyclic array of continuously bonded atoms. It may be viewed as a reaction proceeding through a fully conjugated cyclic transition state. The number of atoms in the cyclic array is usually six, but other numbers are also possible. The term embraces a variety of processes, including cycloadditions, cheletropic reactions, electrocyclic reactions and sigmatropic rearrangements, etc. (provided they are concerted). See also multi-centre reaction.
The differentiation between two symmetry-allowed processes, for example the
See More O'Ferrall-Jencks diagram.
The term persistent is used to characterize radicals which have lifetimes of several minutes or greater in dilute solution in inert solvents. Persistence is a kinetic or reactivity property. In contrast, radical stability, which is a thermodynamic property, is expressed in terms of the C-H bond strength of the appropriate hydrocarbon. The lifetime of a radical is profoundly influenced by steric shielding of the radical centre by bulky substituents. GRILLER and INGOLD (1976). See also transient.
A plot of observed rate coefficient, or more usually its decadic logarithm, against pH of solution, other variables being kept constant.
The phenomenon of rate enhancement of a reaction between chemical species located in different phases (immiscible liquids or solid and liquid) by addition of a small quantity of an agent (called the "phase-transfer catalyst") that extracts one of the reactants, most commonly an anion, across the interface into the other phase so that reaction can proceed. These catalysts are salts of "onium ions" (e.g. tetraalkylammonium salts) or agents that complex inorganic cations (e.g. crown ethers). The catalyst cation is not consumed in the reaction although an anion exchange does occur.
See bridged carbocation.
The cleavage of one or more covalent bonds in a molecular entity resulting from absorption of light, or a photochemical process in which such cleavage is an essential part. For example:
The term is used incorrectly to describe irradiation of a sample, although in the combination flash photolysis this usage is accepted. IUPAC PHOTOCHEMICAL GLOSSARY (1992).
For a reactant molecule RY, the polar effect of the group R comprises all the processes whereby a substituent may modify the electrostatic forces operating at the reaction centre Y, relative to the standard RoY. These forces may be governed by charge separations arising from differences in the electronegativity of atoms (leading to the presence of dipoles), the presence of unipoles, or electron delocalization. It is synonymous with electronic effect or "electrical effect" of a substituent as distinguished from other substituent effects, e.g. steric effects.
Sometimes, however, the term "polar effect" is taken to refer to the influence, other than steric, that non-conjugated substituents exert on reaction rates, i.e. effects connected with electron delocalization between a substituent and the molecular framework to which it is attached are excluded. Polar effect is then not synonymous with electronic effect. See also field effect, inductive effect, mesomeric effect.
See polarity.
When applied to solvents, this rather ill-defined term covers their overall solvation capability (solvation power) for solutes (i.e. in chemical equilibria: reactants and products; in reaction rates: reactants and activated complex; in light absorptions: ions or molecules in the ground and excited state), which in turn depends on the action of all possible, nonspecific and specific, intermolecular interactions between solute ions or molecules and solvent molecules, excluding such interactions leading to definite chemical alterations of the ions or molecules of the solute. Occasionally, the term solvent polarity is restricted to nonspecific solute/solvent interactions only (i.e. to van der Waals forces). See also Dimroth-Reichardt ET parameter, Grunwald-Winstein equation, ionizing power, Kamlet-Taft solvent parameters, van der Waals forces, Z-value. REICHARDT (1965, 1988).
The ease of distortion of the electron cloud of a molecular entity by an electric field (such as that due to the proximity of a charged reagent). It is experimentally measured as the ratio of induced dipole moment (uind) to the field E which induces it:
![[alpha]](../greek/ALPHA.GIF) =
= ![[mu]](../greek/MU.GIF) ind/E
ind/E
The units of are
See ambident.
A curve describing the variation of the potential energy of the system of atoms that make up the reactants and products of a reaction as a function of one geometric coordinate, and corresponding to the "energetically easiest passage" from reactants to products (i.e. along the line produced by joining the paths of steepest descent from the transition state to the reactants and to the products). For an elementary reaction the relevant geometric coordinate is the reaction coordinate; for a stepwise reaction it is the succession of reaction coordinates for the successive individual reaction steps. (The reaction coordinate is sometimes approximated by a quasi-chemical index of reaction progress, such as "degree of atom transfer" or bond order of some specified bond.) See also potential-energy (reaction) surface, Gibbs energy diagram.
potential-energy (reaction) surface
A geometric hypersurface on which the potential energy of a set of reactants is plotted as a function of the coordinates representing the molecular geometries of the system.
For simple systems two such coordinates (characterizing two variables that change during the progress from reactants to products) can be selected, and the potential energy plotted as a contour map.
For simple elementary reactions, e.g. ![]() A + B-C,
A + B-C,
For more complicated reactions a different choice of two coordinates is sometimes preferred, e.g. the bond orders of two different bonds. Such a diagram is often arranged so that reactants are located at the bottom left corner and products at the top right. If the trace of the representative point characterizing the route from reactants to products follows two adjacent edges of the diagram, the changes represented by the two coordinates take place in distinct succession; if the trace leaves the edges and crosses the interior of the diagram, the two changes are concerted. In many qualitative applications it is convenient (although not strictly equivalent) for the third coordinate to represent the standard Gibbs energy rather than potential energy.
Using bond orders is, however, an oversimplification, since these are not well-defined, even for the transition state. (Some reservations concerning the diagrammatic use of Gibbs energies are noted under Gibbs energy diagram.)
The energetically easiest route from reactants to products on the potential-energy contour map defines the potential-energy profile. ALBERY (1967); MORE O'FERRALL (1970); JENCKS (1972, 1985). See also reaction coordinate.
A step on the reaction path of some stepwise reactions in which the molecular entity C is already present in an encounter pair or encounter complex with A during the formation of B from A, e.g.
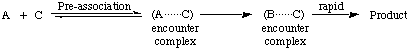
In this mechanism the chemical species C may but does not necessarily assist the formation of B from A, which may itself be a bimolecular reaction with some other reagent.
Pre-association is important when B is too short-lived to permit B and C to come together by diffusion. See also microscopic diffusion control, spectator mechanism.
See encounter complex.
pre-equilibrium (or prior equilibrium)
A rapidly reversible step preceding the rate-limiting step in a stepwise reaction. For example
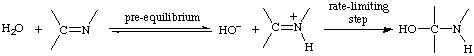
See also kinetic equivalence.
See energy of activation, entropy of activation.
primary kinetic electrolyte effect
See kinetic electrolyte effect.
primary kinetic isotope effect
See isotope effect.
One of the conceptually simpler molecular changes into which an elementary reaction can be notionally dissected. Such changes include bond rupture, bond formation, internal rotation, change of bond length or bond angle, bond migration, redistribution of charge, etc.
The concept of primitive changes is helpful in the detailed verbal description of elementary reactions, but a primitive change does not represent a process that is by itself necessarily observable as a component of an elementary reaction. IUPAC REACTION MECHANISMS (1989).
See pre-equilibrium.
The step of a stepwise reaction, in which the product distribution is determined. The product-determining step may be identical to, or occur later than, the rate-controlling step on the reaction coordinate.
The term is used for reactions under kinetic control where the selectivity parallels the relative (thermodynamic) stabilities of the products. Product development control is usually associated with a transition state occurring late on the reaction coordinate. See also steric-approach control, thermodynamic control.
See pseudo-catalysis.
See chain reaction.
See protogenic.
Capable of acting as a proton (hydron) donor strongly or weakly acidic (as a Brønsted acid). The term is preferred to the synonym "protic" or the more ambiguous expression "acidic" by itself. Also called HBD (hydrogen bond donor) solvent. See protophilic solvent.
This term has been used synonymously with proton (hydron)-transfer reaction. Because of its misleading similarity to hydrolysis, photolysis, etc., its use is discouraged. See also autoprotolysis.
The negative of the enthalpy change in the gas phase reaction (real or hypothetical) between a proton (more appropriately hydron) and the chemical species concerned, usually an electrically neutral species to give the conjugate acid of that species. Proton affinity is often, but unofficially, abbreviated as PA. LIAS, LIEBMAN, and LEVIN (1984). See also gas phase basicity.
A chemical reaction, the main feature of which is the intermolecular or intramolecular transfer of a proton (hydron) from one binding site to another. For example,
In the detailed description of proton transfer reactions, especially of rapid proton transfers between electronegative atoms, it should always be specified whether the term is used to refer to the overall process (including the more-or-less encounter-controlled formation of a hydrogen bonded complex and the separation of the products; see microscopic diffusion control) or just to the proton transfer event (including solvent rearrangement) by itself. See also autoprotolysis, tautomerism.
Capable of acting as proton acceptor, strongly or weakly basic (as a Brønsted base). Also called HBA (hydrogen bond acceptor) solvent. See also protogenic solvent.
prototropic rearrangement (or prototropy)
See tautomerism.
If an acid or base is present in nearly constant concentration throughout a reaction in solution (owing to buffering or the use of a large excess), it may be found to increase the rate of that reaction and also to be consumed during the process. The acid or base is then not a catalyst and the phenomenon cannot be called catalysis according to the well-established meaning of these terms in chemical kinetics, although the mechanism of such a process is often intimately related to that of a catalysed reaction. It is recommended that the term pseudo-catalysis be used in these and analogous cases (not necessarily involving acids or bases). For example, if a Brønsted acid accelerates the hydrolysis of an ester to a carboxylic acid and an alcohol, this is properly called acid catalysis, whereas the acceleration, by the same acid, of hydrolysis of an amide should be described as pseudo-catalysis by the acid: the "acid pseudo-catalyst" is consumed during the reaction through formation of an ammonium ion. The terms "general acid pseudo-catalysis" and "general base pseudo-catalysis" may be used as the analogues of general acid catalysis and general base catalysis.
The term "base-promoted", "base-accelerated, or "base-induced" is sometimes used for reactions that are pseudo-catalysed by bases. However, the term "promotion" also has a different meaning in other chemical contexts.
pseudo-first order rate coefficient
See order of reaction.
+ pseudomolecular rearrangement
The use of this awkwardly formed term is discouraged. It is synonymous with "intermolecular rearrangement".
A concerted transformation is pseudopericyclic if the primary changes in bonding occur within a cyclic array of atoms at one (or more) of which nonbonding and bonding atomic orbitals interchange roles.
A formal example is the enol
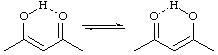
Because the pi and sigma atomic orbitals that interchange roles are orthogonal, such a reaction does not proceed through a fully conjugated transition state and is thus not a pericyclic reaction and therefore not governed by the rules that express orbital symmetry restrictions applicable to pericyclic reactions. ROSS, SEIDERS and LEMAL (1976).
A term sometimes used as synonymous with pseudo-first order, but is inherently meaningless. See molecularity, order of reaction.
Thermolysis, usually associated with exposure to a high temperature. See also flash vacuum pyrolysis.
An adduct formed by electron-pair donation from a pi orbital into a sigma orbital, or from a sigma orbital into a pi orbital, or from a pi orbital into a pi orbital. For example:
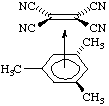
Such an adduct has commonly been known as a "pi complex", but, as the bonding is not necessarily weak, it is better to avoid the term complex, in accordance with the recommendations in this Glossary. See also coordination.
See sigma, pi.
+ -complex
See -adduct.
-electron acceptor, -electron donor group
A substituent capable of a +R (e.g. NO2) or -R (e.g. OCH3) effect, respectively. See electronic effect, polar effect, ![[sigma]](../greek/SIGMA.GIF) -constant.
-constant.
See sigma, pi.
quantitative structure-activity relationships (QSAR)
The building of structure-biological activity models by using regression analysis with physicochemical constants, indicator variables or theoretical calculations. The term has been extended by some authors to include chemical reactivity, i.e. activity is regarded as synonymous with reactivity. This extension is, however, discouraged. See CHARTON (1989). See also correlation analysis.
The number of defined events which occur per photon absorbed by the system. The integral quantum yield is
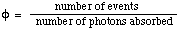
For a photochemical reaction,

The differential quantum yield is
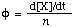
where d[X]/dt is the rate of change of the amount of (substance) concentration of a measurable quantity, and n the amount of photons (mol or its equivalent einsteins) absorbed per unit time. ![[phi]](../greek/PHI.GIF) can be used for photophysical processes or photochemical reactions. IUPAC PHOTOCHEMICAL GLOSSARY (1992).
can be used for photophysical processes or photochemical reactions. IUPAC PHOTOCHEMICAL GLOSSARY (1992).
ALBERY, W. J. (1967), Prog. Reaction Kinetics, 4, 353-398.
GRILLER, D., and INGOLD, K. U. (1976), Acc. Chem. Res., 9, 13-19.
JENCKS, W. P. (1972), Chem. Rev., 72, 705-718.
JENCKS, W. P. (1985), Chem. Rev., 85, 511-527.
LIAS, S. G., LIEBMAN, J. F., and LEVIN, R. P. (1984), J. Phys. Chem. Ref. Data, 13, 695-808.
MORE O'FERRALL, R. A. (1970), J. Chem. Soc. (B), 274-277.
REICHARDT, C. (1965), Angew. Chem., Int. Ed. Engl., 4, 29-40.
ROSS, J. A., SEIDERS, R. P., and LEMAL, D. M. (1976), J. Am. Chem. Soc., 98, 4325-4327.
STOCK, L. M., and BROWN, H. C. (1963), Adv. Phys. Org. Chem., 1, 35-154.
TAFT, R. W., Jr., and TOPSOM, R. D. (1987), Progr. Phys. Org. Chem., 16, 1-83.
Return to home page for Glossary of terms used in Physical Organic Chemistry.