Contents
cage; cage compound; canonical form; captodative effect; carbanion; carbene; carbenium centre; carbenium ion; carbenoid; carbocation; carbonium ion; carbyne; catalysis; catalysis law; catalyst; catalytic coefficient; cationotropic rearrangement (cationotropy); cation radical; chain reaction; chain transfer; charge density; charge population; charge-transfer complex; chelation; cheletropic reaction; chelotropic reaction; chemical flux; chemically induced dynamic nuclear polarization (CIDNP); chemical reaction; chemical relaxation; chemical shift (NMR); chemical species; chemoselective, chemoselectivity; + chemospecificity; chromophore; CIDNP (Chemically Induced Dynamic Nuclear Polarization); cine-substitution; class (a) metal ion; class (b) metal ion; clathrate
An aggregate of molecules, generally in the condensed phase, that surrounds the fragments formed, for example, by thermal or photochemical dissociation. Because the cage hinders the separation of the fragments by diffusion, they may preferentially react with one another ("cage effect") but not necessarily to re-form the precursor species. For example,
See also geminate recombination.
A polycyclic compound having the shape of a cage (see OLAH, 1990). The term is also used for inclusion compounds.
Effect on the stability of a carbon centred radical determined by the combined action of a captor (electron withdrawing) and a dative (electron releasing) substituent, both attached to the radical centre. The term is also used for certain unsaturated compounds. BORDWELL and LYNCH (1989); SUSTMANN and KORTH (1990); VIEHE, JANOUSEK, MERENY, and STELLA (1985).
Generic name for anions containing an even number of electrons and having an unshared pair of electrons on a tervalent carbon atom (e.g. Cl3C- or HC![[triple bond]](../greek/TRIPLEB.GIF) C-) or - if the ion is mesomeric (see mesomerism) - having at least one significant contributing structure with an unshared pair of electrons on a tervalent carbon atom, for example,
C-) or - if the ion is mesomeric (see mesomerism) - having at least one significant contributing structure with an unshared pair of electrons on a tervalent carbon atom, for example,
![[resonance arrow]](../greek/RESARR.GIF) CH3C(=O)-CH--C(=O)CH3
CH3C(=O)-CH--C(=O)CH3See also radical ion.
Generic name for the species H2C: and substitution derivatives thereof, containing an electrically neutral bivalent carbon atom with two nonbonding electrons. The nonbonding electrons may have antiparallel spins (singlet state) or parallel spins (triplet state). Use of the alternative name "methylene" as a generic term is not recommended. See also biradical.
The three-coordinate carbon atom in a carbenium ion to which the excess positive charge of the ion (other than that located on heteroatoms) may be formally considered to be largely attributed, i.e., which has one vacant p-orbital. (N.B. It is not always possible to uniquely identify such an atom.) This formal attribution of charge often does not express the real charge distribution.
A generic name for carbocations, real or hypothetical, that have at least one important contributing structure containing a tervalent carbon atom with a vacant p-orbital. (The name implies a protonated carbene or a substitution derivative thereof.)
The term was proposed (and rejected) as a replacement for the traditional usage of the name carbonium ion.
To avoid ambiguity, the name should not be used as the root for the systematic nomenclature of carbocations. The corresponding difficulty confused carbonium ion nomenclature for many years. For example, the term "ethylcarbonium ion" has at times been used to refer either to CH3CH2+ (ethyl cation) or (correctly) to CH3CH2CH2+ (propyl cation). For the nomenclature of carbenium ions see IUPAC NOMENCLATURE GUIDE (1993).
A carbene like chemical species but with properties and reactivity differing from the free carbene itself, e.g.
A cation containing an even number of electrons with a significant portion of the excess positive charge located on one or more carbon atoms. This is a general term embracing carbenium ions, all types of carbonium ions, vinyl cations, etc. Carbocations may be named by adding the word "cation" to the name of the corresponding radical [IUPAC ORGANIC RULES (1979), IUPAC NOMENCLATURE GUIDE (1993).] Such names do not imply structure (e.g. whether three-coordinated or five-coordinated carbon atoms are present). OLAH and SCHLEYER (1972). See also bridged carbocation, radical ion.
The term should be used with great care since several incompatible meanings are currently in use. It is not acceptable as the root for systematic nomenclature for carbocations.
(1) In most of the existing literature the term is used in its traditional sense for what is here defined as carbenium ion.
(2) A carbocation, real or hypothetical, that contains at least one five-coordinate carbon atom.
(3) A carbocation, real or hypothetical, whose structure cannot adequately be described by two-electron two-centre bonds only. (The structure may involve carbon atoms with a coordination number greater than five.)
Generic name for the species HC![[3 dots]](../greek/3DOT.GIF) and substitution derivatives thereof, such as EtO2C-C containing an electrically neutral univalent carbon atom with three non-bonding electrons. Use of the alternative name "methylidyne" as a generic term is not recommended.
and substitution derivatives thereof, such as EtO2C-C containing an electrically neutral univalent carbon atom with three non-bonding electrons. Use of the alternative name "methylidyne" as a generic term is not recommended.
The action of a catalyst.
See Brønsted relation.
A substance that participates in a particular chemical reaction and thereby increases its rate but without a net change in the amount of that substance in the system. At the molecular level, the catalyst is used and regenerated during each set of microscopic chemical events leading from a molecular entity of reactant to a molecular entity of product. See also autocatalytic reaction, bifunctional catalysis, catalytic coefficient, electron-transfer catalysis, general acid catalysis, general base catalysis, intramolecular catalysis, micellar catalysis, Michaelis-Menten kinetics, phase-transfer catalysis, pseudo-catalysis, rate of reaction, specific catalysis.
If the rate of reaction (v) is expressible in the form
![[Sigma]](../greek/SSIGMA.GIF) ki[Ci]ni) [A]
ki[Ci]ni) [A]![[alpha]](../greek/ALPHA.GIF) [B]
[B]![[beta]](../greek/BETA.GIF) ...
...
where A, B, ... are reactants and Ci represents one of a set of catalysts, then the proportionality factor ki is the catalytic coefficient of the particular catalyst Ci. Normally the partial order of reaction (ni) with respect to a catalyst will be unity, so that ki is an (++...+1)th order rate coefficient. The proportionality factor k0 is the (++...)th order rate coefficient of the uncatalysed component of the total reaction.
cationotropic rearrangement (cationotropy)
See tautomerism.
See radical ion.
A reaction in which one or more reactive reaction intermediates (frequently radicals) are continuously regenerated, usually through a repetitive cycle of elementary steps (the "propagation step"). For example, in the chlorination of methane by a radical mechanism, Cl. is continuously regenerated in the chain propagation steps:
H3C. + Cl2 ![]() CH3Cl + Cl.
CH3Cl + Cl.
In chain polymerization reactions, reactive intermediates of the same types, generated in successive steps or cycles of steps, differ in relative molecular mass, as in
See also chain transfer, initiation, termination.
The abstraction, by the radical end of a growing chain-polymer, of an atom from another molecule. The growth of the polymer chain is thereby terminated but a new radical, capable of chain propagation and polymerization, is simultaneously created. For the example of alkene polymerization cited for a chain reaction, the reaction
represents a chain transfer, the radical Cl3C. inducing further polymerization:
Cl3CCH2C.HPh + H2C=CHPh ![]() Cl3CCH2CHPhCH2C.HPh
Cl3CCH2CHPhCH2C.HPh
The phenomenon occurs also in other chain reactions such as cationic polymerization. See also telomerization.
See electron density.
The net electric charge on a specified atom in a molecular entity, as determined by some prescribed definition, e.g. that of MULLIKEN (1955). See also electron density.
A ground state adduct which exhibits an observable charge transfer absorption band. IUPAC PHOTOCHEMICAL GLOSSARY (1992).
The formation or presence of bonds (or other attractive interactions) between two or more separate binding sites within the same ligand and a single central atom. A molecular entity in which there is chelation (and the corresponding chemical species) is called a "chelate". The terms bidentate (or didentate), tridentate, tetradentate... multidentate are used to indicate the number of potential binding sites of the ligand, at least two of which must be used by the ligand in forming a "chelate". For example, the bidentate ethylenediamine forms a chelate with CuI in which both nitrogen atoms of ethylenediamine are bonded to copper. (The use of the term is often restricted to metallic central atoms.)
The phrase "separate binding sites" is intended to exclude cases such as [PtCl3(CH2=CH2)]-, ferrocene, and (benzene)tricarbonylchromium in which ethene, the cyclopentadienyl group, and benzene, respectively, are considered to present single binding sites to the respective metal atom, and which are not normally thought of as chelates (see hapto). See also cryptand.
A form of cycloaddition across the terminal atoms of a fully conjugated system with formation of two new sigma bonds to a single atom of the ("monocentric") reagent. There is formal loss of one pi bond in the substrate and an increase in coordination number of the relevant atom of the reagent. An example is the addition of sulfur dioxide to butadiene:
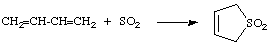
The reverse of this type of reaction is designated "cheletropic elimination". WOODWARD and HOFFMANN (1969).
Alternative (and etymologically more correct) name for cheletropic reaction. DEWAR (1971).
A concept related to rate of reaction, particularly applicable to the progress in one direction only of component reaction steps in a complex system or to the progress in one direction of reactions in a system at dynamic equilibrium (in which there are no observable concentration changes with time). Chemical flux (![[phi]](../greek/PHI.GIF) ) is a derivative with respect to time, and has the dimensions of amount of substance per unit volume transformed per unit time.
) is a derivative with respect to time, and has the dimensions of amount of substance per unit volume transformed per unit time.
The sum of all the chemical fluxes leading to destruction of B is designated the "total chemical flux out of B" (symbol -B); the corresponding formation of B by concurrent elementary reactions is the "total chemical flux into B or A" (symbol B).
For the mechanism
![[reversable arrow]](POgif/CHEM11.GIF) C
C
C + D ![[arrow]](POgif/CHEM2.GIF) E
E
the total chemical flux into C is caused by the single reaction (1):
C = 1whereas the chemical flux out of C is a sum over all reactions that remove C:
-C = -1 + 2
where -1 is the "chemical flux out of C into B (and/or A)" and 2 is the "chemical flux out of C into E". The rate of appearance of C is then given by
C - -C
In this system 1 (or -A) can be regarded as the hypothetical rate of decrease in the concentration of A due to the single (unidirectional) reaction (1) proceeding in the assumed absence of all other reactions.
For a non-reversible reaction
![[arrow]](POgif/CHEM1.GIF) P
P
-d[A]/dt = 1
If two substances A and P are in chemical equilibrium,
![[reversable arrow]](../greek/REVARR.GIF) P
Pthen
A = -A = P = -Pand
GOLD (1979). See also order of reaction, rate-limiting step, steady state.
chemically induced dynamic nuclear polarization (CIDNP)
See CIDNP.
A process that results in the interconversion of chemical species. Chemical reactions may be elementary reactions or stepwise reactions. (It should be noted that this definition includes experimentally observable interconversions of conformers.)
Detectable chemical reactions normally involve sets of molecular entities, as indicated by this definition, but it is often conceptually convenient to use the term also for changes involving single molecular entities (i.e. "microscopic chemical events"). See also identity reaction.
If the equilibrium mixture of a chemical reaction is disturbed by a sudden change, especially of some external parameter (such as temperature, pressure, or electrical field strength), the system will readjust itself to a new position of the chemical equilibrium or return to the original position, if the perturbation is temporary. The readjustment is known as chemical relaxation.
In many cases, and in particular when the displacement from equilibrium is slight, the progress of the system towards equilibrium can be expressed as a first-order law
![[tau]](../greek/TAU.GIF) )
)where (Ceq)1 and (Ceq)2 are the equilibrium concentrations of one of the chemical species involved in the reaction before and after the change in the external parameter, and Ct is its concentration at time t. The time parameter t, named relaxation time, is related to the rate constants of the chemical reaction involved.
Measurements of the relaxation times by relaxation methods [involving a temperature jump (T-jump), pressure jump, electric field jump or a periodic disturbance of an external parameter, as in ultrasonic techniques] are commonly used to follow the kinetics of very fast reactions. See BERNASCONI (1976); LEFFLER and GRUNWALD (1963); see also relaxation.
chemical shift (NMR), ![[delta]](../greek/DELTA.GIF) (SI unit: 1)
(SI unit: 1)
The variation of the resonance frequency of a nucleus in nuclear magnetic resonance (NMR) spectroscopy in consequence of its magnetic environment. The chemical shift of a nucleus, , is expressed in ppm by its frequency, ![[nu]](../greek/NU.GIF) cpd, relative to a standard, ref, and defined as
cpd, relative to a standard, ref, and defined as
= 106(cpd - ref)/o
where o is the operating frequency of the spectrometer. For 1H and 13C NMR the reference signal is usually that of tetramethylsilane (SiMe4). Other references are used in the older literature and in other solvents, such as D2O.
If a resonance signal occurs at lower frequency or higher applied field than an arbitrarily selected reference signal, it is said to be upfield, and if resonance occurs at higher frequency or lower applied field, the signal is downfield. Resonance lines upfield from SiMe4 have positive, and resonance lines downfield from SiMe4 have negative -values.
An ensemble of chemically identical molecular entities that can explore the same set of molecular energy levels on the time scale of the experiment. The term is applied equally to a set of chemically identical atomic or molecular structural units in a solid array.
For example, two conformational isomers may be interconverted sufficiently slowly to be detectable by separate NMR spectra and hence to be considered to be separate chemical species on a time scale governed by the radiofrequency of the spectrometer used. On the other hand, in a slow chemical reaction the same mixture of conformers may behave as a single chemical species, i.e. there is virtually complete equilibrium population of the total set of molecular energy levels belonging to the two conformers.
Except where the context requires otherwise, the term is taken to refer to a set of molecular entities containing isotopes in their natural abundance.
The wording of the definition given in the first paragraph is intended to embrace both cases such as graphite, sodium chloride, or a surface oxide, where the basic structural units may not be capable of isolated existence, as well as those cases where they are.
In common chemical usage, and in this Glossary, generic and specific chemical names (such as radical or hydroxide ion) or chemical formulae refer either to a chemical species or to a molecular entity.
chemoselective, chemoselectivity
Chemoselectivity is the preferential reaction of a chemical reagent with one of two or more different functional groups. A reagent has a high chemoselectivity if reaction occurs with only a limited number of different functional groups. For example, sodium tetrahydroborate is a more chemoselective reducing agent than is lithium tetrahydroaluminate. The concept has not been defined in more quantitative terms. The term is also applied to reacting molecules or intermediates which exhibit selectivity towards chemically different reagents.
Some authors use the term chemospecificity for 100% chemoselectivity. However, this usage is discouraged. See TROST (1980). See also regioselectivity, stereoselectivity, stereospecificity.
See chemoselectivity.
The part (atom or group of atoms) of a molecular entity in which the electronic transition responsible for a given spectral band is approximately localized. The term arose in the dyestuff industry, referring originally to the groupings in the molecule that are responsible for the dye's colour. WITT (1876).
CIDNP (Chemically Induced Dynamic Nuclear Polarization)
Non-Boltzmann nuclear spin state distribution produced in thermal or photochemical reactions, usually from colligation and diffusion, or disproportionation of radical pairs, and detected by NMR spectroscopy by enhanced absorption or emission signals.
A substitution reaction (generally aromatic) in which the entering group takes up a position adjacent to that occupied by the leaving group. For example,
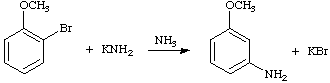
See also tele-substitution.
A metal ion that combines preferentially with ligands containing ligating atoms that are the lightest of their Periodic Group. See also class (b) metal ion, hard acid.
A metal ion that combines preferentially with ligands containing ligating atoms other than the lightest of their Periodic Group.
See also class (a) metal ion, hard acid.
See host, inclusion compound.
BERNASCONI, C. F. (1976), "Relaxation Kinetics", Academic Press, New York.
BERNASCONI, C. F. (1992), Adv. Phys. Org. Chem., 27, 119-238.
BORDWELL, F. G., and LYNCH, T.-Y. (1989), J. Am. Chem. Soc., 111, 7558-7562.
DEWAR, M. J. S. (1971), Angew. Chem., Int. Ed. Engl., 10, 761-776.
GOLD, V. (1979), Nouveau J. Chim., 3, 69-71.
*IUPAC NOMENCLATURE GUIDE (1993). IUPAC: Organic Chemistry Division: Commission on Organic Nomenclature. Guide to IUPAC Nomenclature of Organic Compounds, Blackwell, Oxford, 1993.
MULLIKEN, R. S. (1955), J. Chem. Phys., 23, 1833-1840, 1841-1846, 2338-2342, 2343-2346.
OLAH, G. A. (Ed.) (1990), "Cage Hydrocarbons". Wiley, New York.
OLAH, G. A., and SCHLEYER, P. v. R. (1972), "Carbonium Ions", Vols I-V, Wiley, New York.
SUSTMANN, R., and KORTH, H.-G. (1990), Adv. Phys. Org. Chem., 26, 131-178.
TROST, B. M. (1980), Acc. Chem. Res., 13, 385-393.
VIEHE, H. G., JANOUSEK, Z., MERËNY, R., and STELLA, L. (1985), Acc. Chem. Res., 18, 148-154.
WITT, O. N. (1876), Ber. Deut. Chem. Ges., 9, 522-527.
WOODWARD, R. B., and HOFFMANN, R. (1969), Angew. Chem., Int. Ed. Engl., 8, 781-853.
Return to home page for Glossary of terms used in Physical Organic Chemistry.