![[rho]](../greek/RHO.GIF) -value (rho-value);
-value (rho-value); ![[sigma]](../greek/SIGMA.GIF) -equation (rho-sigma equation)
-equation (rho-sigma equation)
Contents
radical (or free radical); radical anion; radical centre(s); radical combination; radical ion; radical pair (or geminate pair); radiolysis; rate coefficient; rate constant; rate-controlling step; rate-determining step, rate-limiting step; rate law (or empirical differential rate equation); rate of appearance; rate of reaction; reacting bond rules; reaction; reaction coordinate; reaction mechanism; reaction path; reaction stage; reaction step; reactive, reactivity; reactivity index; reactivity-selectivity principle (RSP); rearrangement; rearrangement stage; reduction; reductive elimination; regioselectivity, regioselective; relaxation; reorganization energy; resonance; resonance effect; resonance energy; + retroaddition; + retrocycloaddition; retro-ene reaction; reverse micelle (or reversed micelle); Ritchie equation; -value (rho-value); -equation (rho-sigma equation)
A molecular entity such as .CH3, .SnH3, Cl. possessing an unpaired electron. (In these formulae the dot, symbolizing the unpaired electron, should be placed so as to indicate the atom of highest spin density, if this is possible.) Paramagnetic metal ions are not normally regarded as radicals. However, in the "isolobal analogy" (see HOFFMANN (1982)), the similarity between certain paramagnetic metal ions and radicals becomes apparent.
At least in the context of physical organic chemistry, it seems desirable to cease using the adjective "free" in the general name of this type of chemical species and molecular entity, so that the term "free radical" may in future be restricted to those radicals which do not form parts of radical pairs.
Depending upon the core atom that possesses the unpaired electron, the radicals can be described as carbon-, oxygen-, nitrogen-, metal-centred radicals. If the unpaired electron occupies an orbital having considerable s or more or less pure p character, the respective radicals are termed sigma or pi radicals.
In the past, the term "radical" was used to designate a substituent group bound to a molecular entity, as opposed to "free radical", which nowadays is simply called radical. The bound entities may be called groups or substituents, but should no longer be called radicals. IUPAC NOMENCLATURE GUIDE (1993). See also biradical.
See radical ion.
The atom (or group of atoms) in a polyatomic radical on which an unpaired electron is largely localized. Attachment of a monovalent atom to a radical centre gives a molecule for which it is possible to write a Lewis formula in which the normal stable valencies are assigned to all atoms.
See colligation.
A radical that carries an electric charge. A positively charged radical is called a "radical cation" (e.g., the benzene radical cation C6H6.+); a negatively charged radical is called a "radical anion" (e.g., the benzene radical anion
Unless the positions of unpaired spin and charge can be associated with specific atoms, superscript dot and charge designations should be placed in the order .+ or . - suggested by the name "radical ion", (e.g C3H6.+).
Note: In the previous edition of the Glossary, it was recommended to place the charge designation directly above the centrally placed dot. However, this format is now discouraged because of the difficulty of extending it to ions bearing more than one charge, and/or more than one unpaired electron.
In mass spectroscopic usage the symbol for the charge precedes the dot representing the unpaired electron. IUPAC MASS SPECTROMETRY (1991).
radical pair (or geminate pair)
The term is used to identify two radicals in close proximity in solution, within a solvent cage. They may be formed simultaneously by some unimolecular process, e.g., peroxide decomposition, or they may have come together by diffusion. While the radicals are together, correlation of the unpaired electron spins of the two species cannot be ignored: this correlation is responsible for the CIDNP phenomenon. See also geminate recombination.
The cleavage of one or several bonds resulting from exposure to high-energy radiation. The term is also often used loosely to specify the method of irradiation ("pulse radiolysis") used in any radiochemical reaction, not necessarily one involving bond cleavage.
See order of reaction, kinetic equivalence.
rate constant, k (SI unit: s-1 (dm3 mol-1)n-1)
See order of reaction.
A rate-controlling (rate-determining or rate-limiting) step in a reaction occurring by a composite reaction sequence is an elementary reaction the rate constant for which exerts a strong effect - stronger than that of any other rate constant - on the overall rate. It is recommended that the expressions rate-controlling, rate-determining and rate-limiting be regarded as synonymous, but some special meanings sometimes given to the last two expressions are considered under a separate heading.
A rate-controlling step can be formally defined on the basis of a control function (or control factor) CF, identified for an elementary reaction having a rate constant ki by
where v is the overall rate of reaction. In performing the partial differentiation all equilibrium constants Kj and all rate constants except ki are held constant. The elementary reaction having the largest control factor exerts the strongest influence on the rate v, and a step having a CF much larger than any other step may be said to be rate-controlling.
A rate-controlling step defined in the way recommended here has the advantage that it is directly related to the interpretation of kinetic isotope effects. IUPAC CHEMICAL KINETICS (1991)
As formulated this implies that all rate constants are of the same dimensionality. Consider however the reaction of A and B to give an intermediate C, which then reacts further with D to give products:
C + D
Assuming that C reaches a steady state, then the observed rate is given by
Considering k2[D] a pseudo-first order rate constant, then k2[D] >> k-1, and the observed rate v = k1[A][B] and kobs = k1
Step (1) is said to be the rate-controlling step.
If k2[D] << k-1, then the observed rate
where K is the equilibrium constant for the pre-equilibrium (1) and is equal to k1/k-1, and kobs = Kk2.
Step (2) is said to be the rate-controlling step.
See also Gibbs energy diagram, microscopic diffusion control, mixing control, rate-determining step, rate-limiting step.
rate-determining step, rate-limiting step
These terms are best regarded as synonymous with rate-controlling step. However, other meanings that have been given to them should be mentioned, as it is necessary to be aware of them in order to avoid confusion:
Sometimes the term rate-determining is used as a special case of rate-controlling, being assigned only to an initial slow step which is followed by rapid steps. Such a step imposes an upper limit on the rate, and has also been called rate-limiting.
In view of the considerable danger of confusion when special meanings are applied to rate-determining and rate-limiting, it is recommended that they be regarded as synonymous, with the meaning explained under the entry rate-controlling step. See Michaelis-Menten kinetics. IUPAC CHEMICAL KINETICS (1991).
rate law (or empirical differential rate equation)
An expression for the rate of reaction of a particular reaction in terms of concentrations of chemical species and constant parameters (normally rate coefficients and partial orders of reaction) only. For examples of rate laws see equations (1) to (3) under kinetic equivalence, and (1) under steady state.
See rate of reaction.
For the general chemical reaction
occurring under constant-volume conditions, without an appreciable build-up of reaction intermediates, the rate of reaction v is defined as
where symbols placed inside square brackets denote amount (or amount of substance) concentrations (conventionally expressed in units of mol dm-3). The symbols R and r are also commonly used in place of v. It is recommended that the unit of time should always be the second.
In such a case the rate of reaction differs from the rate of increase of concentration of a product P by a constant factor (the reciprocal of its coefficient in the stoichiometric equation, p) and from the rate of decrease of concentration of the reactant A by 1/a. [It should be noted that all coefficients in the stoichiometric equation are positive; those for products (p, q...) will therefore differ in sign from the stoichiometric numbers defined in IUPAC MANUAL (1979).]
The quantity
defined by the equation
(where nA designates the amount of substance A, conventionally expressed in units of mole) may be called the "rate of conversion" and is appropriate when the use of concentrations is inconvenient, e.g. under conditions of varying volume. In a system of constant volume, the rate of reaction is equal to the rate of conversion per unit volume throughout the reaction.
For a stepwise reaction this definition of "rate of reaction" (and "extent of reaction", ![[partial differential]](../greek/PARDIF.GIF) lnv/lnki)Kj,kj
lnv/lnki)Kj,kj![[reversible arrow rates k1 & k-1]](POgif/RATE6.GIF) C (1)
C (1)![[arrow rate k2]](POgif/RATE7.GIF) Products (2)
Products (2)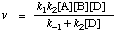
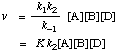
![]() pP + qQ ...
pP + qQ ...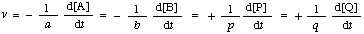
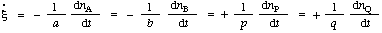
![[xi]](../greek/PI.GIF) ) will apply only if there is no accumulation of intermediate or formation of side products. It is therefore recommended that the term "rate of reaction" be used only in cases where it is experimentally established that these conditions apply. More generally, it is recommended that, instead, the terms "rate of disappearance" or "rate of consumption" of A (i.e. -d[A]/dt, the rate of decrease of concentration of A) or "rate of appearance" of P (i.e. d[P]/dt, the rate of increase of concentration of product P) be used, depending on the concentration change of the particular chemical species
) will apply only if there is no accumulation of intermediate or formation of side products. It is therefore recommended that the term "rate of reaction" be used only in cases where it is experimentally established that these conditions apply. More generally, it is recommended that, instead, the terms "rate of disappearance" or "rate of consumption" of A (i.e. -d[A]/dt, the rate of decrease of concentration of A) or "rate of appearance" of P (i.e. d[P]/dt, the rate of increase of concentration of product P) be used, depending on the concentration change of the particular chemical species
The symbol v (without lettered subscript) should be used only for rate of reaction; v with a lettered subscript (e.g. vA) refers to a rate of appearance or rate of disappearance (e.g. of the chemical species A).
N.B. This definition is consistent with CODATA (1974) recommendations and with IUPAC MANUAL APPENDIX V (1981), but differs from the unconventional terminology in the earlier IUPAC MANUAL (1979). See also chemical relaxation, lifetime, order of reaction.
(1) For an internal motion of a molecular entity corresponding to progress over a transition state (energy maximum), any change that makes the motion more difficult will lead to a new molecular geometry at the energy maximum, in which the motion has proceeded further. Changes that make the motion less difficult will have the opposite effect. (This rule corresponds to the Hammond principle.
(2) For an internal motion of a molecular entity that corresponds to a vibration, any change that tends to modify the equilibrium point of the vibration in a particular direction will actually shift the equilibrium in that direction.
(3) Effects on reacting bonds (bonds made or broken in the reaction) are the most significant. The bonds nearest the site of structural change are those most strongly affected.
THORNTON (1967). See also More O'Ferrall-Jencks diagram.
See chemical reaction.
A geometric parameter that changes during the conversion of one (or more) reactant molecular entities into one (or more) product molecular entities and whose value can be taken for a measure of the progress of an elementary reaction (for example, a bond length or bond angle or a combination of bond lengths and/or bond angles; it is sometimes approximated by a non-geometric parameter, such as the bond order of some specified bond).
In the formalism of "transition-state theory", the reaction coordinate is that coordinate in a set of curvilinear coordinates obtained from the conventional ones for the reactants which, for each reaction step, leads smoothly from the configuration of the reactants through that of the transition state to the configuration of the products. The reaction coordinate is typically chosen to follow the path along the gradient (path of shallowest ascent/deepest descent) of potential energy from reactants to products.
The term has also been used interchangeably with the term transition coordinate, applicable to the coordinate in the immediate vicinity of the potential energy maximum. Being more specific, the name transition coordinate is to be preferred in that context. MARCUS (1966). See also potential-energy profile, potential-energy reaction surface.
See mechanism.
(1) A synonym for mechanism.
(2) A trajectory on the potential-energy surface.
(3) A sequence of synthetic steps.
A set of one or more (possibly experimentally inseparable) reaction steps leading to and/or from a detectable or presumed reaction intermediate.
An elementary reaction, constituting one of the stages of a stepwise reaction in which a reaction intermediate (or, for the first step, the reactants) is converted into the next reaction intermediate (or, for the last step, the products) in the sequence of intermediates between reactants and products. See also rate-limiting step, reaction stage.
As applied to a chemical species, the term expresses a kinetic property. A species is said to be more reactive or to have a higher reactivity in some given context than some other (reference) species if it has a larger rate constant for a specified elementary reaction. The term has meaning only by reference to some explicitly stated or implicitly assumed set of conditions. It is not to be used for reactions or reaction patterns of compounds in general.
The term is also more loosely used as a phenomenological description not restricted to elementary reactions. When applied in this sense the property under consideration may reflect not only rate, but also equilibrium, constants. See also stable, unreactive, unstable.
Any numerical index derived from quantum mechanical model calculations that permits the prediction of relative reactivities of different molecular sites. Many indices are in use, based on a variety of theories and relating to various types of reaction. The more successful applications have been to the substitution reactions of conjugated systems where relative reactivities are determined largely by changes of pi-electron energy.
reactivity-selectivity principle (RSP)
This idea may be expressed loosely as: the more reactive a reagent is, the less selective it is.
Consider two substrates S1 and S2 undergoing the same type of reaction with two reagents R1 and R2, S2 being more reactive than S1, and R2 more reactive than R1 in the given type of reaction. The relative reactivities (in log units, see selectivity) for the four possible reactions may notionally be represented as follows:

With the positions of (S1 + R1), (S2 + R1), and (S1 + R2) fixed, there are three types of positions for (S2 + R2):
In position (i) the selectivity of R2 for the two substrates, measured by a is the same as the selectivity of R1 for the two substrates, also a.
In position (ii) the selectivity of R2 for the two substrates, measured by b, is less than the selectivity of R1 for the two substrates, i.e. b < a. It is this situation which is in accord with the RSP.
In position (iii) the selectivity of R2 for the two substrates, measured by c, is greater than the selectivity of R1 for the two substrates, i.e. c > a. This situation may be described as anti-RSP.
There are many examples in which the RSP is followed, but there are also many examples corresponding to situations (i) and (iii). The RSP is in accord with intuitive feeling and certainly holds in the limiting case when reactivity is controlled by diffusion. However, the validity of the RSP is a matter of great controversy "...and diverse opinions have been expressed, from declaring the reactivity-selectivity principle as a universal law up to 'virtually useless in practice as a general rule'." [EXNER (1988)]. ARGILE, CAREY, FUKATA, HARCOURT, MORE O'FERRAL AND MURPHY (1985); BUNCEL and WILSON (1987); JOHNSON (1975); STOCK and BROWN (1963).
See degenerate rearrangement, molecular rearrangement, sigmatropic rearrangement.
The elementary reaction or reaction stage (of a molecular rearrangement) in which there is both making and breaking of bonds between atoms common to a reactant and a reaction product or a reaction intermediate. If the rearrangement stage consists of a single elementary reaction, this is a "rearrangement step".
The complete transfer of one or more electrons to a molecular entity (also called "electronation"), and, more generally, the reverse of the processes described under oxidation (2), (3).
The reverse of oxidative addition.
regioselectivity, regioselective
A regioselective reaction is one in which one direction of bond making or breaking occurs preferentially over all other possible directions. Reactions are termed completely (100%) regioselective if the discrimination is complete, or partially (x%), if the product of reaction at one site predominates over the product of reaction at other sites. The discrimination may also semi-quantitatively be referred to as high or low regioselectivity.
(Originally the term was restricted to addition reactions of unsymmetrical reagents to unsymmetrical alkenes.)
In the past, the term "regiospecificity" was proposed for 100% regioselectivity. This terminology is not recommended owing to inconsistency with the terms stereoselectivity and stereospecificity . See ADAMS (1992); HASSNER (1968). See also chemoselectivity.
Passage of an excited or otherwise perturbed system towards or into thermal equilibrium with its environment. IUPAC PHOTOCHEMICAL GLOSSARY (1988). See also chemical relaxation.
In a one-electron transfer reaction
![[reversible arrow]](../greek/REVARR.GIF) A. - + D.+
A. - + D.+
the reorganization energy ![[lambda]](../greek/LAMBDA.GIF) is the energy required for all structural adjustments (in the reactants and in the surrounding solvent molecules) which are needed in order that A and D assume the configuration required for the transfer of the electron. See intrinsic barrier, Marcus equation.
is the energy required for all structural adjustments (in the reactants and in the surrounding solvent molecules) which are needed in order that A and D assume the configuration required for the transfer of the electron. See intrinsic barrier, Marcus equation.
In the context of chemistry, the term refers to the representation of the electronic structure of a molecular entity in terms of contributing structures. Resonance among contributing structures means that the wavefunction is represented by "mixing" the wavefunctions of the contributing structures. The concept is the basis of the quantum mechanical valence bond methods. The resulting stabilization is linked to the quantum mechanical concept of "resonance energy". The term resonance is also used to refer to the delocalization phenomenon itself. See ATKINS (1974). See also mesomerism.
This is the term most commonly used to describe the influence (on reactivity, spectra, etc.) of a substituent through electron delocalization into or from the substituent. The use of the term obviates the need to attempt to distinguish between the operation of the mesomeric effect and the electromeric effect. (An alternative term with essentially the same meaning is "conjugative effect". At one time "tautomeric effect" was also used, but was abandoned because tautomerism implies reorganization of the atomic nuclei.) The effect is symbolized by R.
The difference in potential energy between the actual molecular entity and the contributing structure of lowest potential energy. The resonance energy cannot be measured, but only estimated, since contributing structures are not observable molecular entities. See resonance.
See cycloelimination.
See cycloelimination.
See ene reaction.
reverse micelle (or reversed micelle)
See inverted micelle.
The linear free energy relation
applied to the reactions between nucleophiles and certain large and relatively stable organic cations, e.g. arenediazonium, triarylmethyl, and aryltropylium cations in various solvents. kN is the rate constant for reaction of a given cation with a given nucleophilic system (i.e. given nucleophile in a given solvent). k0 is the rate constant for the same cation with water in water, and N+ is a parameter which is characteristic of the nucleophilic system and independent of the cation. A surprising feature of the equation is the absence of a coefficient of N+, characteristic of the substrate (cf. the s in the Swain-Scott equation), even though values of N+ vary over 13 log units. The equation thus involves a gigantic breakdown of the reactivity-selectivity principle. The equation has been extended both in form and in range of application. RITCHIE (1972, 1978, 1986). See also CHAPMAN and SHORTER (1978).
A measure of the susceptibility to the influence of substituent groups on the rate constant or equilibrium constant of a particular organic reaction involving a family of related substrates. Defined by Hammett for the effect of ring substituents in meta- and para-positions of aromatic side-chain reactions by the empirical "-equation" of the general form
X
in which x is a constant characteristic of the substituent X and of its position in the reactant molecule.
More generally (and not only for aromatic series), -values (modified with appropriate subscripts and superscripts) are used to designate the susceptibility of reaction series for families of various organic compounds to any substituent effects, as given by the modified set of -constants in an empirical -correlation.
Reactions with a positive -value are accelerated (or the equilibrium constants of analogous equilibria are increased) by substituents with positive -constants. Since the sign of was defined so that substituents with a positive increase the acidity of benzoic acid, such substituents are generally described as attracting electrons away from the aromatic ring. It follows that reactions with a positive -value are considered to involve a transition state (or reaction product) so that the difference in energy between this state and the reactants is decreased by a reduction in electron density at the reactive site of the substrate. See also Hammett equation, -constant, Taft equation.
-equation (rho-sigma equation)
See Hammett equation, -value, -constant, Taft equation.
ADAMS, D. L. (1992), J. Chem. Educ., 69, 451-452.
ATKINS, P. W. (1974), "Quanta: a Handbook of Concepts", Clarendon Press, Oxford.
BUNCEL, E., and WILSON, H. (1987), J. Chem. Educ., 64, 475-480.
CODATA (1974), Codata Bulletin No. 13, Dec. 1974, p. 4.
EXNER, O. (1988), "Correlation Analysis of Chemical Data", Plenum, New York, SNTL, Prague.
HASSNER, A. (1968), J. Org. Chem., 33, 2684-2686.
HOFFMANN, R. (1982), Angew. Chem., Int. Ed. Engl., 21, 711-724.
*IUPAC NOMENCLATURE GUIDE (1993). IUPAC: Organic Chemistry Division: Commission on Organic Nomenclature. Guide to IUPAC Nomenclature of Organic Compounds, Blackwell, Oxford, 1993.
JOHNSON, C. D. (1975), Chem. Rev., 75, 755-765.
MARCUS, R. A. (1966), J. Chem. Phys., 45, 4493-4499, 4500-4504.
RITCHIE, C. D. (1972), Acc. Chem. Res., 5, 348-354.
RITCHIE, C. D. (1978), Pure Appl Chem., 50, 1281-1290.
RITCHIE, C. D. (1986), Can. J. Chem., 64, 2239-2250.
STOCK, L. M., and BROWN, H. C. (1963), Adv. Phys. Org. Chem., 1, 35-154.
THORNTON, E. R., (1967), J. Am. Chem. Soc., 89, 2915-2927.
Return to home page for Glossary of terms used in Physical Organic Chemistry.