GLOSSARY OF TERMS USED IN PHYSICAL ORGANIC CHEMISTRY
(IUPAC Recommendations 1994)
I
Continued from terms starting with H
Contents
identity reaction; imbalance; + imene; + imidogen; + imidonium ion; + imin; + imine radical; inclusion compound (or inclusion complex); induction period; inductive effect; inductomeric effect; inert; inhibition; initiation; inner-sphere (electron transfer); insertion; intermediate; intermolecular; internal return; intimate ion pair; intramolecular; intramolecular catalysis; intrinsic barrier; inverse kinetic isotope effect; inverted micelle; ionic strength; ionization; ionization energy; ionizing power; ion pair; ion pair return; ipso-attack; isodesmic reaction; isoelectronic; isoentropic; isoequilibrium relationship; isokinetic relationship; isolobal; isomer; isomerization; isosbestic point; isoselective relationship; isotope effect; isotope effect, equilibrium; isotope effect, heavy atom; isotope effect, intramolecular; isotope effect, inverse; isotope effect, kinetic; isotope effect, primary; isotope effect, secondary; isotope effect, solvent; isotope effect, steric; isotope effect, thermodynamic; isotope exchange; isotopic perturbation, method of; isotopic scrambling; isotopologue; isotopomer; isovalent hyperconjugation
identity reaction
A chemical reaction whose products are chemically identical with the reactants, for example the bimolecular self exchange reaction of CH3I with I-. See also degenerate rearrangement.
imbalance
The situation in which reaction parameters that characterize different bond forming or bond breaking processes in the same reaction have developed to different extents as the transition state is approached along some arbitrarily defined reaction coordinate. For example, in the nitroalkane anomaly, the Brønsted ![[beta]](../greek/BETA.GIF) exponent for proton removal is smaller than the Brønsted
exponent for proton removal is smaller than the Brønsted ![[alpha]](../greek/ALPHA.GIF) for the nitroalkane, because of imbalance between the amount of bond breaking and resonance delocalization in the transition state. Imbalance is common in reactions such as elimination, addition and other complex reactions that involve proton (hydron) transfer. BERNASCONI (1992). See also synchronous, synchronization (principle of imperfect synchronization).
for the nitroalkane, because of imbalance between the amount of bond breaking and resonance delocalization in the transition state. Imbalance is common in reactions such as elimination, addition and other complex reactions that involve proton (hydron) transfer. BERNASCONI (1992). See also synchronous, synchronization (principle of imperfect synchronization).
+ imene
See nitrene.
+ imidogen
See nitrene.
+ imidonium ion
See nitrenium ion.
+ imin
See nitrene.
+ imine radical
See nitrene.
inclusion compound (or inclusion complex)
A complex in which one component (the host) forms a cavity or, in the case of a crystal, a crystal lattice containing spaces in the shape of long tunnels or channels in which molecular entities of a second chemical species (the guest) are located. There is no covalent bonding between guest and host, the attraction being generally due to van der Waals forces. If the spaces in the host lattice are enclosed on all sides so that the guest species is "trapped" as in a cage, such compounds are known as "clathrates" or "cage" compounds".
induction period
The initial slow phase of a chemical reaction which later accelerates. Induction periods are often observed with radical reactions, but they may also occur in other systems (for example before steady-state concentration of the reactants is reached).
inductive effect
In strict definition, an experimentally observable effect (on rates of reaction, etc.) of the transmission of charge through a chain of atoms by electrostatic induction. A theoretical distinction may be made between the field effect, and the inductive effect as models for the Coulomb interaction between a given site within a molecular entity and a remote unipole or dipole within the same entity. The experimental distinction between the two effects has proved difficult, except for molecules of peculiar geometry, which may exhibit "reversed field effects". Ordinarily the inductive effect and the field effect are influenced in the same direction by structural changes in the molecule and the distinction between them is not clear. This situation has led many authors to include the field effect in the term "inductive effect". Thus the separation of ![[sigma]](../greek/SIGMA.GIF) values into inductive and resonance components does not imply the exclusive operation of a through-bonds route for the transmission of the non-conjugative part of the substituent effect. To indicate the all-inclusive use of the term inductive, the phrase "so-called inductive effect" is sometimes used. Certain modern theoretical approaches suggest that the "so-called inductive effect" reflects a field effect rather than through-bonds transmission. EHRENSON, BROWNLEE and TAFT (1973); TAFT and TOPSOM (1987). See also field effect, mesomeric effect, polar effect.
values into inductive and resonance components does not imply the exclusive operation of a through-bonds route for the transmission of the non-conjugative part of the substituent effect. To indicate the all-inclusive use of the term inductive, the phrase "so-called inductive effect" is sometimes used. Certain modern theoretical approaches suggest that the "so-called inductive effect" reflects a field effect rather than through-bonds transmission. EHRENSON, BROWNLEE and TAFT (1973); TAFT and TOPSOM (1987). See also field effect, mesomeric effect, polar effect.
inductomeric effect
A molecular polarizability effect occurring by the inductive mechanism of electron displacement. The consideration of such an effect and the descriptive term have been regarded as obsolescent or even obsolete, but in recent years theoretical approaches have reintroduced substituent polarizability as a factor governing reactivity, etc. and its parametrization has been proposed. See TAFT and TOPSOM (1987); INGOLD (1953).
inert
Stable and unreactive under specified conditions.
inhibition
The decrease in rate of reaction brought about by the addition of a substance (inhibitor), by virtue of its effect on the concentration of a reactant, catalyst or reaction intermediate. For example, molecular oxygen and p-benzoquinone can react as "inhibitors" in many reactions involving radicals as intermediates by virtue of their ability to act as scavengers toward these radicals.
If the rate of a reaction in the absence of inhibitor is vo and that in the presence of a certain amount of inhibitor is v, the degree of inhibition (i) is given by
i = (vo - v)/vo
See also mechanism based inhibition.
initiation
A reaction or process generating free radicals (or some other reactive reaction intermediates) which then induce a chain reaction. For example, in the chlorination of alkanes by a radical mechanism the initiation step is the dissociation of molecular chlorine.
inner-sphere (electron transfer)
Historically an electron transfer between two metal centres sharing a ligand or atom in their respective coordination shells. The definition has more recently been extended to any situation in which the interaction between the donor and acceptor centres in the transition state is significant (>20 kJ mol-1). IUPAC PHOTOCHEMICAL GLOSSARY (1992). See also outer-sphere electron transfer.
insertion
A chemical reaction or transformation of the general type
X-Z + Y ![[arrow]](../greek/ARROW.GIF) X-Y-Z
X-Y-Z
in which the connecting atom or group Y replaces the bond joining the parts X and Z of the reactant XZ. An example is the carbene insertion reaction
R3C-H + H2C: R3C-CH3
The reverse of an insertion is called an extrusion. See also -addition.
intermediate
A molecular entity with a lifetime appreciably longer than a molecular vibration (corresponding to a local potential energy minimum of depth greater than RT) that is formed (directly or indirectly) from the reactants and reacts further to give (either directly or indirectly) the products of a chemical reaction; also the corresponding chemical species. See reaction step, elementary reaction, stepwise reaction.
intermolecular
(1) Descriptive of any process that involves a transfer (of atoms, groups, electrons, etc.) or interactions between two or more molecular entities.
(2) Relating to a comparison between different molecular entities.
See also intramolecular.
internal return
See ion-pair return.
intimate ion pair
See ion pair.
intramolecular
(1) Descriptive of any process that involves a transfer (of atoms, groups, electrons, etc.) or interactions between different parts of the same molecular entity.
(2) Relating to a comparison between atoms or groups within the same molecular entity.
See also intermolecular.
intramolecular catalysis
The acceleration of a chemical transformation at one site of a molecular entity through the involvement of another functional ("catalytic") group in the same molecular entity, without that group appearing to have undergone change in the reaction product. The use of the term should be restricted to cases for which analogous intermolecular catalysis by chemical species bearing that catalytic group is observable. Intramolecular catalysis can be detected and expressed in quantitative form by a comparison of the reaction rate with that of a comparable model compound in which the catalytic group is absent, or by measurement of the effective molarity of the catalytic group. See also effective molarity, neighbouring group participation.
intrinsic barrier
The Gibbs energy of activation (![[Delta]](../greek/DDELTA.GIF)
![[double dagger]](../greek/DDAGGER.GIF) G) in the limiting case where Go = 0, i.e. when the effect of thermodynamic driving force is eliminated. According to the Marcus equation, the intrinsic barrier is related to the reorganization energy,
G) in the limiting case where Go = 0, i.e. when the effect of thermodynamic driving force is eliminated. According to the Marcus equation, the intrinsic barrier is related to the reorganization energy, ![[lambda]](../greek/LAMBDA.GIF) , of the reaction by the equation
, of the reaction by the equation
G = /4
CANNON (1980); SCHLESENER, AMATORE and KOCHI (1986).
inverse kinetic isotope effect
See isotope effect.
inverted micelle
The reversible formation of association colloids from surfactants in non-polar solvents leads to aggregates termed inverted (or inverse, reverse or reversed) micelles. Such association is often of the type
Monomer ![[reversible arrow]](../greek/REVARR.GIF) Dimer Trimer ... n-mer
Dimer Trimer ... n-mer
and the phenomenon of critical micelle concentration (or an analogous effect) is consequently not observed.
In an inverted micelle the polar groups of the surfactants are concentrated in the interior and the lipophilic groups extend towards and into the non-polar solvent.
ionic strength, I (SI unit: mol dm-3)
In a solution of fully dissociated electrolytes the ionic strength is defined as I = 0.5![[Sigma]](../greek/SSIGMA.GIF) iciZi2, in which ci is the concentration and Zi the charge number of ionic species i.
iciZi2, in which ci is the concentration and Zi the charge number of ionic species i. ![[mu]](../greek/MU.GIF) is also defined as Im = 0.5imiZi2, where mi is the molality.
is also defined as Im = 0.5imiZi2, where mi is the molality.
ionization
The generation of one or more ions. It may occur, e.g. by loss of an electron from a neutral molecular entity, by the unimolecular heterolysis of such an entity into two or more ions, or by a heterolytic substitution reaction involving neutral molecules, such as
CH3CO2H + H2O H3O+ + CH3CO2-
Ph3CCl + AlCl3 Ph3C+ + AlCl4- (electrophile-assisted)
Ph3CCl Ph3C+ Cl-(ion pair, in benzene)
The loss of an electron from a singly, doubly, etc. charged cation is called second, third, etc. ionization. This terminology is used especially in mass spectroscopy. See also dissociation, ionization energy.
ionization energy, Ei (SI unit kJ mol-1 or J per molecule)
The minimum energy required to remove an electron from an isolated molecular entity (in its vibrational ground state) in the gaseous phase. If the resulting molecular entity is considered to be in its vibrational ground state, one refers to the energy as the "adiabatic ionization energy". If the molecular entity produced possesses the vibrational energy determined by the Franck-Condon principle (according to which the electron ejection takes place without an accompanying change in molecular geometry), the energy is called the "vertical ionization energy". The name ionization energy is preferred to the somewhat misleading earlier name "ionization potential". See also ionization.
ionizing power
A term to denote the tendency of a particular solvent to promote ionization of an uncharged or, less often, charged solute. The term has been used both in a kinetic and in a thermodynamic context. See also Dimroth-Reichardt ET parameter, Grunwald-Winstein equation, Z-value.
ion pair
A pair of oppositely charged ions held together by Coulomb attraction without formation of a covalent bond. Experimentally, an ion pair behaves as one unit in determining conductivity, kinetic behaviour, osmotic properties, etc.
Following Bjerrum, oppositely charged ions with their centres closer together than a distance
q = 8.36 x 106 Z+Z-/(![[epsilon]](../greek/EPSILON.GIF) rT) pm
rT) pm
are considered to constitute an ion pair ("Bjerrum ion pair"). [Z+ and Z- are the charge numbers of the ions, and r is the relative permittivity (or dielectric constant) of the medium.]
An ion pair, the constituent ions of which are in direct contact (and not separated by an intervening solvent or other neutral molecule) is designated as a "tight ion pair" (or "intimate" or "contact ion pair"). A tight ion pair of X+ and Y- is symbolically represented as X+Y-.
By contrast, an ion pair whose constituent ions are separated by one or several solvent or other neutral molecules is described as a "loose ion pair", symbolically represented as X+||Y-. The members of a loose ion pair can readily interchange with other free or loosely paired ions in the solution. This interchange may be detectable (e.g., by isotopic labelling) and thus afford an experimental distinction between tight and loose ion pairs.
A further conceptual distinction has sometimes been made between two types of loose ion pairs. In "solvent-shared ion pairs" the ionic constituents of the pair are separated by only a single solvent molecule, whereas in "solvent-separated ion pairs" more than one solvent molecule intervenes. However, the term "solvent-separated ion pair" must be used and interpreted with care since it has also widely been used as a less specific term for "loose" ion pair. See also common-ion effect, dissociation, ion-pair return, special salt effect.
ion pair return
The recombination of a pair of ions R+ and Z- formed from ionization of RZ.
If the ions are paired as a tight ion pair and recombine without prior separation into a loose ion pair this is called "internal ion-pair return":
R+Z- (tight ion pair) RZ (covalent molecule)
It is a special case of "primary geminate recombination".
If the ions are paired as a loose ion pair and form the covalent chemical species via a tight ion pair, this is called "external ion-pair return":
R+||Z- (loose ion pair) R+Z- (tight ion pair) RZ (covalent molecule)
It is a special case of "secondary geminate recombination".
When the covalent molecule RZ is reformed without direct evidence of prior partial racemization or without other direct evidence of prior formation of a tight ion pair, (e.g., without partial racemization if the group R is suitably chiral) the internal ion-pair return is sometimes called a "hidden return".
External (unimolecular) ion-pair return is to be distinguished from "external (bimolecular) ion return", the (reversible) process whereby dissociated ions are converted into loose ion pairs:
R+ + Z- R+||Z-
ipso-attack
The attachment of an entering group to a position in an aromatic compound already carrying a substituent group (other than hydrogen). The entering group may displace that substituent group but may also itself be expelled or migrate to a different position in a subsequent step. The term "ipso-substitution" is not used, since it is synonymous with substitution.
For example:
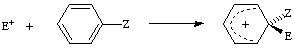
where E+ is an electrophile and Z is a substituent (other than hydrogen). See also cine-substitution, tele-substitution.
isodesmic reaction
A reaction (actual or hypothetical) in which the types of bonds that are made in forming the products are the same as those which are broken in the reactants, e.g.
PhCOOH + p-ClC6H4COO- PhCOO- + p-ClC6H4COOH
ClCH=CH2 + ClCH2CH2Cl CH2=CH2 + Cl2CHCH2Cl
Such processes have advantages for theoretical treatment. The Hammett equation as applied to equilibria (cf. (a)) essentially deals with isodesmic processes.
For the use of isodesmic processes in quantum chemistry, see HEHRE et al. (1970).
isoelectronic
Two or more molecular entities are described as isoelectronic if they have the same number of valence electrons and the same structure, i.e. number and connectivity of atoms, but differ in some of the elements involved. Thus
CO, N2 and NO+ are isoelectronic
CH2=C=O and CH2=N=N are isoelectronic
CH3COCH3 and CH3N=NCH3 have the same number of electrons, but have different structures, hence they are not described as isoelectronic.
isoentropic
A reaction series is said to be isoentropic if the individual reactions of the series have the same standard entropy of activation.
isoequilibrium relationship
A relationship analogous to the isokinetic relationship but applied to equilibrium data. The equation defining the isoequilibrium temperature is
rH - rS = constant
where H and S are enthalpy and entropy of reaction, respectively. See also isokinetic relationship.
isokinetic relationship
When a series of structurally related substrates undergo the same general reaction or when the reaction conditions for a single substrate are changed in a systematic way, the enthalpies and entropies of activation sometimes satisfy the relation
H - S = constant
where the parameter is independent of temperature. This equation (or some equivalent form) is said to represent an "isokinetic relationship". The temperature T = (at which all members of a series obeying the isokinetic relationship react at the same rate) is termed the "isokinetic temperature".
Supposed isokinetic relationships as established by direct correlation of H with S are often spurious and the calculated value of is meaningless, because errors in H lead to compensating errors in S. Satisfactory methods of establishing such relationships have been devised. EXNER (1973); LEFFLER (1955). See also compensation effect, isoequilibrium relationship, isoselective relationship.
isolobal
The term is used to compare molecular fragments with each other and with familiar species from organic chemistry. Two fragments are isolobal if the number, symmetry properties, approximate energy, and shape of the frontier orbitals and the number of electrons in them are similar. See isoelectronic.
isomer
One of several species (or molecular entities) that have the same atomic composition (molecular formula) but different line formulae or different stereochemical formulae and hence different physical and/or chemical properties.
isomerization
A chemical reaction, the principal product of which is isomeric with the principal reactant. An intramolecular isomerization that involves the breaking or making of bonds is a special case of a molecular rearrangement.
Isomerization does not necessarily imply molecular rearrangement (e.g. in the case of the interconversion of conformational isomers).
isosbestic point
This term is usually employed with reference to a set of absorption spectra, plotted on the same chart for a set of solutions in which the sum of the concentrations of two principal absorbing components, A and B, is constant. The curves of absorbance against wavelength (or frequency) for such a set of mixtures often all intersect at one or more points, called isosbestic points.
Isosbestic points are commonly met when electronic spectra are taken (a) on a solution in which a chemical reaction is in progress (in which case the two absorbing components concerned are a reactant and a product, A + B), or (b) on a solution in which the two absorbing components are in equilibrium and their relative proportions are controlled by the concentration of some other component, typically the concentration of hydrogen ions, e.g., an acid-base indicator equilibrium.
A B + H+aq
The effect may also appear (c) in the spectra of a set of solutions of two unrelated non-interacting components having the same total concentration. In all these examples, A (and/or B) may be either a single chemical species or a mixture of chemical species present in invariant proportion.
If A and B are single chemical species, isosbestic points will appear at all wavelengths at which their molar absorption coefficients (formerly called extinction coefficients) are the same. (A more involved identity applies when A and B are mixtures of constant proportion.)
If absorption spectra of the types considered above intersect not at one or more isosbestic points but over progressively changing wavelength, this is prima facie evidence in case (a) for the formation of a reaction intermediate in substantial concentration (A C B), in case (b) for the involvement of a third absorbing species in the equilibrium, e.g.
A B + H+aq C + 2 H+aq
or in case (c) for some interaction of A and B, e.g.,
A + B C
isoselective relationship
A relationship analogous to the isokinetic relationship, but applied to selectivity data of reactions. At the isoselective temperature, the selectivities of the series of reactions following the relationship are identical. GIESE (1984). See also isoequilibrium relationship, isokinetic relationship.
isotope effect
The effect on the rate or equilibrium constant of two reactions that differ only in the isotopic composition of one or more of their otherwise chemically identical components is referred to as a kinetic isotope effect (see isotope effect, kinetic) or a thermodynamic (or equilibrium) isotope effect (see isotope effect, thermodynamic), respectively.
isotope effect, equilibrium
See isotope effect, thermodynamic.
isotope effect, heavy atom
An isotope effect due to isotopes other than those of hydrogen.
isotope effect, intramolecular
A kinetic isotope effect observed when a single substrate, in which the isotopic atoms occupy equivalent reactive positions, reacts to produce a non-statistical distribution of isotopologue products. In such a case the isotope effect will favor the pathway with lower force constants for displacement of the isotopic nuclei in the transition state.
isotope effect, inverse
A kinetic isotope effect which kl/kh < 1, i.e. the heavier substrate reacts more rapidly than the lighter one, as opposed to the more usual "normal" isotope effect, in which kl/kh > 1. The isotope effect will normally be "normal" when the frequency differences between the isotopic transition states are smaller than in the reactants. Conversely, in inverse isotope effect can be taken as evidence for an increase in the corresponding force constants on passing from the reactant to the transition state.
isotope effect, kinetic
The effect of isotopic substitution on a rate constant is referred to as a kinetic isotope effect.
For example in the reaction
A + B C
the effect of isotopic substitution in reactant A is expressed as the ratio of rate constants kl/kh, where the superscripts l and h represent reactions in which the molecules A contain the light and heavy isotopes, respectively.
Within the framework of transition state theory in which the reaction is rewritten as
A + B [TS] C
and with neglect of isotopic mass on tunnelling and the transmission coefficient, kl/kh can be regarded as if it were the equilibrium constant for an isotope exchange reaction between the transition state [TS] and the isotopically substituted reactant A, and calculated from their vibrational frequencies as in the case of a thermodynamic isotope effect (see isotope effect, thermodynamic).
Isotope effects like the above, involving a direct or indirect comparison of the rates of reaction of isotopologues, are called "intermolecular", in contrast to intramolecular isotope effects (see isotope effect, intramolecular), in which a single substrate reacts to produce a non-statistical distribution of isotopologue product molecules. See WOLFSBERG (1972).
isotope effect, primary
A kinetic isotope effect attributable to isotopic substitution of an atom to which a bond is made or broken in the rate-controlling step or in a pre-equilibrium step of a specified reaction is termed a primary isotope effect. The corresponding isotope effect on the equilibrium constant of a reaction in which one or more bonds to isotopic atoms are broken, is called a "primary equilibrium isotope effect". See also isotope effect, secondary.
isotope effect, secondary
A kinetic isotope effect that is attributable to isotopic substitution of an atom to which bonds are neither made nor broken in the rate-controlling step or in a pre-equilibrium step of a specified reaction, and is therefore not a primary isotope effect, is termed a secondary isotope effect. One speaks of , (etc.) secondary isotope effects, where , (etc.) denote the position of isotopic substitution relative to the reaction centre. The corresponding isotope effect on the equilibrium constant of such a reaction is called a "secondary equilibrium isotope effect".
Secondary isotope effects have been discussed in terms of the conventional electronic effects of physical organic chemistry, e.g. induction, hyperconjugation, hybridization, etc., since these properties are determined by the electron distribution, that depends on vibrationally averaged bond lengths and angles which vary slightly with isotopic substitution. While this usage is legitimate, the term "electronic isotope effect" should be avoided, because of the misleading implication that such an effect is electronic rather than vibrational in origin. See also isotope effect, steric.
isotope effect, solvent
A kinetic or equilibrium isotope effect resulting from change in the isotopic composition of the solvent.
isotope effect, steric
A secondary isotope effect attributed to the different vibrational amplitudes of isotopologues. For example, both the mean and mean-square amplitudes of vibrations associated with C-H bonds are greater than those of C-D bonds. The greater effective bulk of molecules containing the former may be manifested by a steric effect on a rate or equilibrium constant.
isotope effect, thermodynamic
The effect of isotopic substitution on an equilibrium constant is referred to as a thermodynamic (or equilibrium) isotope effect.
For example, the effect of isotopic substitution in reactant A that participates in the equilibrium:
A + B C
is the ratio Kl/Kh of the equilibrium constant for the reaction in which A contains the light isotope to that in which it contains the heavy isotope. The ratio can be expressed as the equilibrium constant for the isotopic exchange reaction:
Al + Ch Ah + Cl
in which reactants such as B that are not isotopically substituted do not appear.
The potential energy surfaces of isotopic molecules are identical to a high degree of approximation, so thermodynamic isotope effects can only arise from the effect of isotopic mass on the nuclear motions of the reactants and products, and can be expressed quantitatively in terms of partition function ratios for nuclear motion:
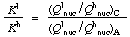
Although the nuclear partition function is a product of the translational, rotational and vibrational partition functions, the isotope effect is determined almost entirely by the last named, specifically by vibrational modes involving motion of isotopically different atoms. In the case of light atoms (i.e. protium vs. deuterium or tritium) at moderate temperatures, the isotope effect is dominated by zero-point energy differences. WOLFSBERG (1972). See also fractionation factor.
isotope exchange
A chemical reaction in which the reactant and product chemical species are chemically identical but have different isotopic composition. In such a reaction the isotope distribution tends towards equilibrium (as expressed by fractionation factors) as a result of transfers of isotopically different atoms or groups. For example,
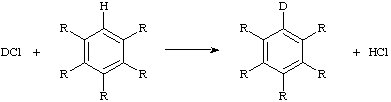
isotopic perturbation, method of
NMR shift difference measurement of the isotope effect on a fast (degenerate) equilibrium between two, except for isotopic substitution, species which are equivalent. This can be used to distinguish a rapidly equilibrating mixture with time-averaged symmetry from a single structure with higher symmetry. SIEHL (1987).
isotopic scrambling
The achievement, or the process of achieving, an equilibrium distribution of isotopes within a specified set of atoms in a chemical species or group of chemical species. For example,
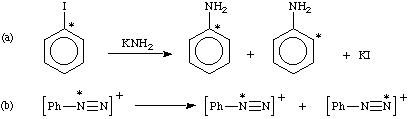
(* denotes position of an isotopically different atom.)
See also fractionation factor.
isotopologue
A molecular entity that differs only in isotopic composition (number of isotopic substitutions), e.g. CH4, CH3D, CH2D2... .
isotopomer
Isomers having the same number of each isotopic atom but differing in their positions. The term is a contraction of "isotopic isomer".
Isotopomers can be either constitutional isomers (e.g. CH2DCH=O and CH3CD=O) or isotopic stereoisomers (e.g. (R)- and (S)-CH3CHDOH or (Z)- and (E)-CH3CH=CHD). IUPAC STEREOCHEMICAL TERMINOLOGY (1993).
isovalent hyperconjugation
See hyperconjugation.
References
BERNASCONI, C. F. (1992), Adv. Phys. Org. Chem., 27, 119-238.
CANNON, R. D. (1980), "Electron Transfer Reactions", Butterworths, London.
EHRENSON, S., BROWNLEE, R. T. C., and TAFT, R. W. (1973), Progr. Phys. Org. Chem., 10, 1-80.
EXNER, O. (1973), Progr. Phys. Org. Chem., 10, 411-482.
GIESE, B. (1984), Acc. Chem. Res., 17, 438-442.
HEHRE, W. J., DITCHFIELD, R., RADOM, L., and POPLE, J. A. (1970), J. Am. Chem. Soc., 92, 4796-4801.
INGOLD, C. K. (1953), "Structure and Mechanism in Organic Chemistry", Cornell University Press, New York.
*IUPAC PHOTOCHEMICAL GLOSSARY (1992). IUPAC: Organic Chemistry Division: Commission on Photochemistry. Glossary of Terms Used in Photochemistry. Draft 1, provisional.
*IUPAC STEREOCHEMICAL TERMINOLOGY (1993). IUPAC: Organic Chemistry Division: Basic Terminology of Stereochemistry. IDCNS and public review. Now published as Basic Terminology of Stereochemistry (IUPAC Recommendations 1996) in Pure Appl. Chem., 68, 2193-2222 (1996).
LEFFLER, J. E. (1955), J. Org. Chem., 20, 1202-1231.
SCHLESENER, C. J., AMATORE, C., and KOCHI, J. K. (1986), J. Phys. Chem., 90, 3747-3756.
SIEHL, H.-U. (1987), Adv. Phys. Org. Chem., 23, 63-163.
TAFT, R. W., Jr., and TOPSOM, R. D. (1987), Progr. Phys. Org. Chem., 16, 1-83.
WOLFSBERG, M. (1972), Acc. Chem. Res., 5, 225-233.
Continue with terms starting with K and L.
Return to home page for Glossary of terms used in Physical Organic Chemistry.