![[alpha]](../greek/ALPHA.GIF) ), hydrogen bond acceptor (
), hydrogen bond acceptor (![[beta]](../greek/BETA.GIF) ), and dipolarity/polarizability (
), and dipolarity/polarizability (![[pi]](../greek/PI.GIF) *) properties of solvents as contributing to overall solvent polarity . KAMLET, ABBOUD and TAFT (1981).
*) properties of solvents as contributing to overall solvent polarity . KAMLET, ABBOUD and TAFT (1981).
Contents
Kamlet-Taft solvent parameters; Kekulé structure (for aromatic compounds); kinetic ambiguity; kinetic control (of product composition); kinetic electrolyte effect (kinetic ionic-strength effect); kinetic equivalence; kinetic isotope effect; Koppel-Palm solvent parameters; Kosower Z-value
labile; least nuclear motion, principle of; leaving group; Leffler's assumption; left-to-right convention; levelling effect; Lewis acid; Lewis acidity; Lewis adduct; Lewis base; Lewis basicity; Lewis formula (electron dot or Lewis structure); lifetime (mean lifetime); ligand; linear free-energy relation; linear Gibbs energy relation; linear solvation energy relationships; line formula; line-shape analysis; Lineweaver-Burk plot; lipophilic; London forces; lone (electron) pair; loose ion pair; LUMO; lyate ion; lyonium ion
Parameters of the Kamlet-Taft solvatochromic relationship which measure separately the hydrogen bond donor (), hydrogen bond acceptor (), and dipolarity/polarizability (*) properties of solvents as contributing to overall solvent polarity . KAMLET, ABBOUD and TAFT (1981).
Kekulé structure (for aromatic compounds)
A representation of an aromatic molecular entity (such as benzene), with fixed alternating single and double bonds, in which interactions between multiple bonds are assumed to be absent.
For benzene,
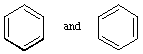
are the Kekulé structures.
Synonymous with kinetic equivalence .
kinetic control (of product composition)
The term characterizes conditions (including reaction times) that lead to reaction products in a proportion governed by the relative rates of the parallel (forward) reactions in which the products are formed, rather than by the respective overall equilibrium constants. See also thermodynamic control .
kinetic electrolyte effect (kinetic ionic-strength effect)
The general effect of an added electrolyte (i.e. an effect other than, or in addition to, that due to its possible involvement as a reactant or catalyst) on the observed rate constant of a reaction in solution. At low concentrations (when only long-range coulombic forces need to be considered) the effect on a given reaction is determined only by the ionic strength of the solution and not by the chemical identity of the ions. For practical purposes, this concentration range is roughly the same as the region of validity of the Debye-Hückel limiting law for activity coefficients. At higher concentrations, the effect of an added electrolyte depends also on the chemical identity of the ions. Such specific action can usually be interpreted as the incursion of a reaction path involving an ion of the electrolyte as reactant or catalyst, in which case the action is not properly to be regarded just as a kinetic electrolyte effect.
Kinetic electrolyte effects are usually (too restrictively and therefore incorrectly) referred to as "kinetic salt effects".
A kinetic electrolyte effect ascribable solely to the influence of the ionic strength on activity coefficients of ionic reactants and transition states is called a "primary kinetic electrolyte effect".
A kinetic electrolyte effect arising from the influence of the ionic strength of the solution upon the pre-equilibrium concentration of an ionic species that is involved in a subsequent rate-limiting step of a reaction is called a "secondary kinetic electrolyte effect". A common case encountered in practice is the effect on the concentration of hydrogen ion (acting as catalyst) produced from the ionization of a weak acid in a buffer solution. See also common-ion effect , order of reaction .
Two reaction schemes are kinetically equivalent if they imply the same rate law .
For example, consider the two schemes (i) and (ii) for the formation of C from A:
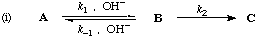
Providing that B does not accumulate as a reaction intermediate.
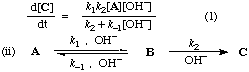
Providing that B does not accumulate as a reaction intermediate
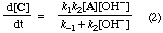
Both equations for d[C]/dt are of the form
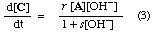
where r and s are constants (sometimes called "coefficients in the rate equation"). The equations are identical in their dependence on concentrations and do not distinguish whether OH- catalyses the formation of B, and necessarily also its reversion to A, or is involved in its further transformation to C. The two schemes are therefore kinetically equivalent under conditions to which the stated provisos apply.
See isotope effect .
Koppel-Palm solvent parameters
Parameters to measure separately the ability of a solvent to enter into non-specific solvent-solute interactions (permittivity ![[epsilon]](../greek/EPSILON.GIF) and refractive index nD) and specific solvent-solute interaction (solvent basicity or nucleophilicity B and solvent acidity or electrophilicity E) as contributing to overall solvent polarity. KOPPEL and PALM (1972).
and refractive index nD) and specific solvent-solute interaction (solvent basicity or nucleophilicity B and solvent acidity or electrophilicity E) as contributing to overall solvent polarity. KOPPEL and PALM (1972).
See Z-value .
The term has loosely been used to describe a relatively unstable and transient chemical species or (less commonly) a relatively stable but reactive species. It must therefore not be used without explanation of the intended meaning. See also inert , persistent , reactive , unreactive .
least nuclear motion, principle of
The hypothesis that, for given reactants, the reactions involving the smallest change in nuclear positions will have the lowest energy of activation . (It is also often simply referred to as principle of least motion.) HINE (1977).
An atom or group (charged or uncharged) that becomes detached from an atom in what is considered to be the residual or main part of the substrate in a specified reaction.
For example, in the heterolytic solvolysis of benzyl bromide in acetic acid
the leaving group is Br- ; in the reaction
the leaving group is NMe3; in the electrophilic nitration of benzene, it is H+. The term has meaning only in relation to a specified reaction. The leaving group is not, in general, the same as the substituent group present in the substrate (e.g. bromo and trimethylammonio in the substrates of the first two examples above.)
A slightly different usage of the term prevails in the (non-mechanistic) naming of transformations, where the actual substituent group present in the substrate (and also in the product) is referred to as the leaving group. See also electrofuge , entering group , nucleofuge .
See Hammond principle .
Arrangement of the structural formulae of the reactants so that the bonds to be made or broken form a linear array in which the electrons move from left to right. IUPAC REACTION MECHANISMS (1989).
The tendency of a solvent to make all Brønsted acids whose acidity exceeds a certain value appear equally acidic. It is due to the complete transfer to a protophilic solvent of a hydron from a dissolved acid stronger than the conjugate acid of the solvent. The only acid present to any significant extent in all such solutions is the lyonium ion . For example, the solvent water has a levelling effect on the acidities of HClO4, HCl, and HI: aqueous solutions of these acids at the same (moderately low) concentrations have the same acidities. A corresponding levelling effect applies to strong bases in protogenic solvents.
A molecular entity (and the corresponding chemical species ) that is an electron-pair acceptor and therefore able to react with a Lewis base to form a Lewis adduct , by sharing the electron pair furnished by the Lewis base. For example:
See also coordination , dipolar bond .
The thermodynamic tendency of a substrate to act as a Lewis acid . Comparative measures of this property are provided by the equilibrium constants for Lewis adduct formation of a series of Lewis acids with a common reference Lewis base . See also acceptor number (AN) , electrophilicity .
The adduct formed between a Lewis acid and a Lewis base .
A molecular entity (and the corresponding chemical species ) able to provide a pair of electrons and thus capable of coordination to a Lewis acid , thereby producing a Lewis adduct .
The thermodynamic tendency of a substance to act as a Lewis base . Comparative measures of this property are provided by the equilibrium constants for Lewis adduct formation for a series of Lewis bases with a common reference Lewis acid . See also donor number (DN) , nucleophilicity .
Lewis formula (electron dot or Lewis structure)
Molecular structure in which the valency electrons are shown as dots so placed between the bonded atoms that one pair of dots represents two electrons or one (single) covalent bond , e.g.
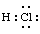
A double bond is represented by two pairs of dots, etc. Dots representing non-bonded outer-shell electrons are placed adjacent to the atoms with which they are associated, but not between the atoms. Formal charges (e.g. +, -, 2+, etc.) are attached to atoms to indicate the difference between the positive nuclear charge (atomic number) and the total number of electrons (including those in the inner shells), on the formal basis that bonding electrons are shared equally between atoms they join. (Bonding pairs of electrons are usually denoted by lines, representing covalent bonds, as in line formulae .)
The lifetime of a chemical species which decays in a first-order process is the time needed for a concentration of this species to decrease to 1/e of its original value. Statistically, it represents the mean life expectancy of an excited species. In a reacting system in which the decrease in concentration of a particular chemical species is governed by a first-order rate law , it is equal to the reciprocal of the sum of the (pseudo)unimolecular rate constants of all processes which cause the decay. When the term is used for processes which are not first order, the lifetime depends on the initial concentration of the species, or of a quencher, and should be called apparent lifetime instead. See also chemical relaxation , half-life , rate of reaction .
If it is possible to indicate a "central atom" in a polyatomic molecular entity , the atoms or groups bound to that atom are called ligands. (Cf. IUPAC INORGANIC NOMENCLATURE (1990); Rule I-10.2.3.) The term is generally used in connection with metallic "central atoms".
In biochemistry a part of a polyatomic molecular entity may be considered central, and atoms, groups or molecules bound to that part are considered ligands (Cf. BIOCHEMICAL NOMENCLATURE (1992)).
A linear correlation between the logarithm of a rate constant or equilibrium constant for one series of reactions and the logarithm of the rate constant or equilibrium constant for a related series of reactions. Typical examples of such relations (also known as linear Gibbs energy relations) are the Brønsted relation , and the Hammett equation (see also ![[sigma]](../greek/SIGMA.GIF) -value ).
-value ).
The name arises because the logarithm of an equilibrium constant (at constant temperature and pressure) is proportional to a standard free energy (Gibbs energy) change, and the logarithm of a rate constant is a linear function of the free energy (Gibbs energy) of activation.
It has been suggested (IUPAC PHYSICAL ORGANIC GLOSSARY (1983)) that this name should be replaced by Linear Gibbs Energy Relation , but at present there is little sign of acceptance of this change.
The area of physical organic chemistry which deals with such relations is commonly referred to as "Linear Free-Energy Relationships".
See linear free-energy relation .
linear solvation energy relationships
Equations involving the application of solvent parameters in linear or multiple (linear) regression expressing the solvent effect on the rate or equilibrium constant of a reaction. See Dimroth-Reichardt ET parameter, Kamlet-Taft solvent parameter , Koppel-Palm solvent parameter , Z-value .
A two-dimensional representation of molecular entities in which atoms are shown joined by lines representing single or multiple bonds, without any indication or implication concerning the spatial direction of bonds. For example, methanol is represented as
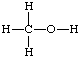
(The term should not be confused with the representation of chemical formulae by the "Wiswesser line notation", a method of string notation. Formulae in this notation are also known as "Wiswesser line formulae".)
Determination of rate constants for a chemical exchange from the shapes of spectroscopic lines of dynamic processes. The method is most often used in nuclear magnetic resonance spectroscopy.
See Michaelis-Menten kinetics .
Literally "fat-loving". Applied to molecular entities (or parts of molecular entities) having a tendency to dissolve in fat-like (e.g. hydrocarbon) solvents. See also hydrophilic , hydrophobic interaction .
Attractive forces between apolar molecules, due to their mutual polarizability. They are also components of the forces between polar molecules. Also called "dispersion forces". See also van der Waals forces .
Two paired electrons localized in the valence shell on a single atom. Lone pairs should be designated with two dots.
The term "nonbonding electron pair" is more appropriate, and is found in many modern text books.
See ion pair.
See frontier orbitals .
The anion produced by hydron removal from a solvent molecule. For example, the hydroxide ion is the lyate ion of water.
The cation produced by hydronation of a solvent molecule. For example, CH3OH2+ is the lyonium ion of methanol. See also onium ion .
BIOCHEMICAL NOMENCLATURE (1992). JCBN/NC-IUBMB Newsletter, in "Biochemical Nomenclature and Related Documents", 2nd edn., Portland Press, London, 335.
HINE, J. (1977), Adv. Phys. Org. Chem., 15, 1-61.
KAMLET, M. J., ABBOUD, J. L. M., and TAFT, R. W. (1981), Progr. Phys. Org. Chem., 13, 485-630.
Return to home page for Glossary of terms used in Physical Organic Chemistry.
![[tau]](../greek/TAU.GIF)