GLOSSARY OF TERMS USED IN PHYSICAL ORGANIC CHEMISTRY
(IUPAC Recommendations 1994)
H
Continued from terms starting with F and G
Contents
half-life; halochromism; Hammett acidity function; Hammett equation (or Hammett relation); Hammond principle (or Hammond postulate); Hansch constant; hapto; hard acid; hard base; heat capacity of activation; + hemileptic; Henderson-Hasselbach equation; heterobimetallic complex; heteroconjugation; heteroleptic; heterolysis, heterolytic; heterolytic bond-dissociation energy; heterovalent hyperconjugation; hidden return; Hildebrand parameter; Hofmann rule; homo; homoaromatic; homoconjugation; homoleptic; homolysis, homolytic; host; Hückel (4n + 2) rule; hybridization; hydration; hydrogen bond; hydrolysis; hydron; hydrophilic; hydrophobic interaction; hyperconjugation; hypsochromic shift
half-life, t1/2 (SI unit: s)
In a kinetic experiment, the time required for the concentration of a particular reacting species to fall to one-half of its initial value. (Its dependence on initial concentration depends upon the order of reaction. It is independent of initial concentration only for a first-order process.) See also lifetime.
halochromism
Halochromism means the colour change which occurs on addition of acid (or base, or a salt) to a solution of a compound. A chemical reaction (e.g. ion formation) transforms a colourless compound into a coloured one. FALBE and REGITZ (1990); REICHARDT, ASHARIN-FARD, and SCHÄFER. (1993).
Hammett acidity function
See acidity function.
Hammett equation (or Hammett relation)
The equation in the form
lg(k/ko) = ![[rho]](../greek/RHO.GIF)
![[sigma]](../greek/SIGMA.GIF)
or
lg(K/Ko) =
applied to the influence of meta- or para-substituents X on the reactivity of the functional group Y in the benzene derivative m- or p-XC6H4Y. k or K is the rate or equilibrium constant, respectively, for the given reaction of m- or p-XC6H4Y; ko or Ko refers to the reaction of C6H5Y, i.e. X = H; is the substituent constant
characteristic of m- or p-X; is the reaction constant characteristic of the given reaction of Y. The equation is often encountered in a form with lg ko or lg Ko written as a separate term on the right hand side, e.g.
lg k = + lg ko
or
lg K = + lg Ko
It then signifies the intercept corresponding to X = H in a regression of lg k or lg K on . HAMMETT (1940, 1970). See also -value, -constant, Taft equation, Yukawa-Tsuno equation.
Hammond principle (or Hammond postulate)
The hypothesis that, when a transition state leading to an unstable reaction intermediate (or product) has nearly the same energy as that intermediate, the two are interconverted with only a small reorganization of molecular structure. Essentially the same idea is sometimes referred to as "Leffler's assumption", namely, that the transition state bears the greater resemblance to the less stable species (reactant or reaction intermediate/product). Many text books and physical organic chemists, however, express the idea in Leffler's form, but attribute it to Hammond.
As a corollary, it follows that a factor stabilising a reaction intermediate will also stabilize the transition state leading to that intermediate.
The acronym "Bemahapothle" (Bell, Marcus, Hammond, Polanyi, Thornton, Leffler) is sometimes used in recognition of the principal contributors towards expansion of the original idea of the Hammond postulate. HAMMOND (1955); LEFFLER (1953); WILLIAMS (1984). See also FARCASIU (1975). See also More O'Ferrall-Jencks diagram.
Hansch constant
A measure of the capability of a solute for hydrophobic (lipophilic) interaction based on the partition coefficient P for distribution of the solute between octan-1-ol and water. The most general way of applying P in correlation analysis, QSAR, etc. is as log P, but the behaviour of substituted benzene derivatives may be quantified by a substituent constant scale, ![[pi]](../greek/PI.GIF) , which is defined in a way analogous to the Hammett scale. There are various scales, depending on the substrate series used as reference. HANSCH and LEO (1979).
, which is defined in a way analogous to the Hammett scale. There are various scales, depending on the substrate series used as reference. HANSCH and LEO (1979).
hapto
The hapto symbol, ![[eta]](../greek/ETA.GIF) with numerical superscript, provides a topological description for the bonding of hydrocarbons and other pi-electron systems to metals, by indicating the connectivity between the ligand and the central atom. For example, 3 indicates that three atoms of the ligand are bonded to the central atom. IUPAC INORGANIC NOMENCLATURE (1990).
with numerical superscript, provides a topological description for the bonding of hydrocarbons and other pi-electron systems to metals, by indicating the connectivity between the ligand and the central atom. For example, 3 indicates that three atoms of the ligand are bonded to the central atom. IUPAC INORGANIC NOMENCLATURE (1990).
hard acid
A Lewis acid with an acceptor centre of low polarizability. Other things being approximately equal, complexes of hard acids and bases or soft acids and bases have an added stabilization (sometimes called "HSAB" rule). For example the hard O- (or N-) bases are preferred to their S- (or P-) analogues by hard acids. Conversely a "soft acid" possesses an acceptor centre of high polarizability and exhibits the reverse preference for coordination of a soft base. These preferences are not defined in a quantitative sense. See PEARSON (1963, 1973); HO (1977). See also class (a) metal ion, hard base.
hard base
A Lewis base with a donor centre (e.g. an oxygen atom) of low polarizability; the converse applies to "soft bases". See also hard acid.
heat capacity of activation, ![[Delta]](../greek/DDELTA.GIF)
![[double dagger]](../greek/DDAGGER.GIF) Cpo (SI unit: J mol-1 K-1)
Cpo (SI unit: J mol-1 K-1)
A quantity related to the temperature coefficient of H (enthalpy of activation) and S (entropy of activation) according to the equations:
Cp = (![[partial differential]](../greek/PARDIF.GIF) H/T)p = T(S/T)p
H/T)p = T(S/T)p
If the rate constant is expressible in the form ln k = a/T + b + c ln T + dT, then
Cp = (c - 1) R + 2dRT
See KOHNSTAM (1967).
+ hemileptic
See homoleptic.
Henderson-Hasselbach equation
An equation of the form
pH = pKa - lg([HA]/[A-])
for the calculation of the pH of solutions where the ratio [HA]/[A-] is known.
heterobimetallic complex
A metal complex having two different metal atoms.
heteroconjugation
+(1) Association between a base and the conjugate acid of a different base through a hydrogen bond (B'....HB+ or A'H ....A-). The term has its origin in the conjugate acid-base pair and is in no way related to conjugation of orbitals. Heteroassociation is a more appropriate term.
+(2) Some authors refer to conjugated systems containing a heteroatom, e.g. pyridine, as "heteroconjugated systems". This usage is discouraged since it inappropriately suggests an analogy to homoconjugation (2), and conflicts with the currently accepted definition of that term.
heteroleptic
Transition metal or Main Group compounds having more than one type of ligand. See also homoleptic.
heterolysis, heterolytic
The cleavage of a covalent bond so that both bonding electrons remain with one of the two fragments between which the bond is broken, e.g.

Heterolytic bond fission is a feature of many bimolecular reactions in solution (e.g., electrophilic substitution, nucleophilic substitution). See also homolysis, heterolytic bond-dissociation energy.
heterolytic bond-dissociation energy
The energy required to break a given bond of some specific compound by heterolysis. For the dissociation of a neutral molecule AB in the gas phase into A+ and B- the heterolytic bond-dissociation energy D(A+B-) is the sum of the bond dissociation energy, D(A-B), and the adiabatic ionization energy of the radical A. minus the electron affinity of the radical B..
heterovalent hyperconjugation
See hyperconjugation.
hidden return
See ion-pair return.
Hildebrand parameter
A parameter measuring the cohesion of a solvent (energy required to create a cavity in the solvent). CHASTRETTE, RAJZMANN, CHANON, and PURCELL (1985).
Hofmann rule
"The principal alkene formed in the decomposition of quaternary ammonium hydroxides that contain different primary alkyl groups is always ethylene, if an ethyl group is present." Originally given in this limited form by A.W. Hofmann, the rule has since been extended and modified as follows: "When two or more alkenes can be produced in a ![[beta]](../greek/BETA.GIF) -elimination reaction, the alkene having the smallest number of alkyl groups attached to the double bond carbon atoms will be the predominant product." This orientation described by the Hofmann rule is observed in elimination reactions of quaternary ammonium salts and tertiary sulfonium salts, and in certain other cases. HOFMANN (1851). See also Saytzeff rule.
-elimination reaction, the alkene having the smallest number of alkyl groups attached to the double bond carbon atoms will be the predominant product." This orientation described by the Hofmann rule is observed in elimination reactions of quaternary ammonium salts and tertiary sulfonium salts, and in certain other cases. HOFMANN (1851). See also Saytzeff rule.
homo
(1) An acronym for Highest Occupied Molecular Orbital. See frontier orbitals.
(2) A prefix (consisting of lower case letters, homo,) used to indicate a higher homologue of a compound.
homoaromatic
Whereas in an aromatic molecule there is continuous overlap of p-orbitals over a cyclic array of atoms, in a homoaromatic molecule there is a formal discontinuity in this overlap resulting from the presence of a single sp3 hybridized atom at one or several positions within the ring; p-orbital overlap apparently bridges these sp3 centres, and features associated with aromaticity are manifest in the properties of the compound. Pronounced homoaromaticity is not normally associated with neutral molecules, but mainly with species bearing an electrical charge, e.g., the "homotropylium" cation, C8H9+,
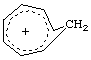
In bis, tris, (etc.) homoaromatic species, two, three, (etc.) single sp3 centres separately interrupt the pi-electron system. See also homoconjugation (2).
homoconjugation
+ (1) Association between a base and its conjugate acid through a hydrogen bond (B...HB+ or AH...A-). Homoassociation is a more appropriate term for this phenomenon.
(2) The orbital overlap of two pi systems separated by a non-conjugating group, such as CH2.
See also conjugate acid-base pair, conjugated system, homoaromatic.
homoleptic
Transition metal or Main Group compounds having only one type of ligand are said to be homoleptic, e.g. TaMe5. See also heteroleptic.
homolysis, homolytic
The cleavage of a bond ("homolytic cleavage" or "homolytic fission") so that each of the molecular fragments between which the bond is broken retains one of the bonding electrons. A unimolecular reaction involving homolysis of a bond (not forming part of a cyclic structure) in a molecular entity containing an even number of (paired) electrons results in the formation of two radicals:

It is the reverse of colligation. Homolysis is also commonly a feature of bimolecular substitution reactions (and of other reactions) involving radicals and molecules. See also bond dissociation energy, heterolysis.
host
A molecular entity that forms complexes with organic or inorganic guests, or a chemical species that can accommodate guests within cavities of its crystal structure. Examples include cryptands and crowns (where there are ion-dipole attractions between heteroatoms and positive ions), hydrogen-bonded molecules that form "clathrates" (e.g. hydroquinone and water), and host molecules of inclusion compounds (e.g. urea or thiourea). van der Waals forces and hydrophobic interactions bind the guest to the host molecule in clathrates and inclusion compounds.
Hückel (4n + 2) rule
Monocyclic planar (or almost planar) systems of trigonally (or sometimes digonally) hybridized atoms that contain (4n + 2) electrons (where n is a non-negative integer) will exhibit aromatic character. The rule is generally limited to n = 0-5.
This rule is derived from the Hückel MO calculation on planar monocyclic conjugated hydrocarbons (CH)m where m is an integer equal to or greater than 3 according to which (4n + 2) electrons are contained in a closed-shell system. Examples of systems that obey the Hückel rule include:
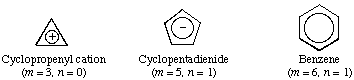
Systems containing 4n electrons (such as cyclobutadiene and the cyclopentadienyl cation) are "antiaromatic". See also conjugation, Möbius aromaticity.
hybridization
Linear combination of atomic orbitals on an atom. Hybrid orbitals are often used in organic chemistry to describe the bonding molecules containing tetrahedral (sp3), trigonal (sp2), and digonal (sp) atoms.
hydration
Addition of water or of the elements of water (i.e. H and OH) to a molecular entity. For example, hydration of ethene:
CH2=CH2 + H2O ![[arrow]](../greek/ARROW.GIF) CH3CH2OH
CH3CH2OH
The term is also used in a more restricted sense for the process:
A (gas) A (aqueous solution)
cf. the use of the term in inorganic/physical chemistry to describe the state of the ions of an electrolyte in aqueous solution. AHRLAND (1979). See also aquation, solvation.
hydrogen bond
The hydrogen bond is a form of association between an electronegative atom and a hydrogen atom attached to a second, relatively electronegative atom. It is best considered as an electrostatic interaction, heightened by the small size of hydrogen, which permits proximity of the interacting dipoles or charges. Both electronegative atoms are usually (but not necessarily) from the first row of the Periodic Table, i.e., N, O, or F. Hydrogen bonds may be intermolecular or intramolecular. With a few exceptions, usually involving fluorine, the associated energies are less than 20-25 kJ mol-1 (5-6 kcal mol-1).
hydrolysis
Solvolysis by water.
hydron
General name for the ion H+ either in natural abundance, or where it is not desired to distinguish between the isotopes, as opposed to proton for 1H+, deuteron for 2H+ and triton for 3H+. IUPAC NAMES FOR HYDROGEN ATOMS (1988).
hydrophilic
"Water loving". The capacity of a molecular entity or of a substituent to interact with polar solvents, in particular with water, or with other polar groups.
hydrophobic interaction
The tendency of hydrocarbons (or of lipophilic hydrocarbon-like groups in solutes) to form intermolecular aggregates in an aqueous medium, and analogous intramolecular interactions. The name arises from the attribution of the phenomenon to the apparent repulsion between water and hydrocarbons. However, the phenomenon ought to be attributed to the effect of the hydrocarbon-like groups on the water-water interaction. The misleading alternative term "hydrophobic bond" is discouraged.
hyperconjugation
In the formalism that separates bonds into and types, hyperconjugation is the interaction of -bonds (e.g. C-H, C-C, etc.) with a network. This interaction is customarily illustrated by contributing structures, e.g. for toluene (below), sometimes said to be an example of "heterovalent" or "sacrificial hyperconjugation", so named because the contributing structure contains one two-electron bond less than the normal Lewis formula for toluene
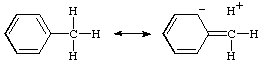
At present, there is no evidence for sacrificial hyperconjugation in neutral hydrocarbons.
The concept of hyperconjugation is also applied to carbenium ions and radicals, where the interaction is now between -bonds and an unfilled or partially filled or p-orbital. A contributing structure illustrating this for the tert-butyl cation is:
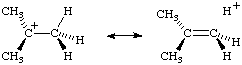
This latter example is sometimes called an example of "isovalent hyperconjugation" (the contributing structure containing the same number of two-electron bonds as the normal Lewis formula).
Both structures shown on the right hand side are also examples of "double bond- no-bond resonance".
The interaction between filled or p orbitals and adjacent antibonding * orbitals is referred to as "negative hyperconjugation", as for example in the fluoroethyl anion:
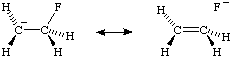
RADOM (1982). See also sigma pi (, ), n-* delocalization.
hypsochromic shift
Shift of a spectral band to higher frequency or shorter wavelength upon substitution or change in medium. It is informally referred to as blue shift. IUPAC PHOTOCHEMICAL GLOSSARY (1988). See also bathochromic shift.
References
AHRLAND, S. (1979), Pure Appl. Chem., 51, 2019-2039.
CHASTRETTE, M., RAJZMANN, M., CHANON, M., and PURCELL, K. F. (1985), J. Am. Chem. Soc., 107, 1-11.
FALBE, J., REGITZ, M. (Eds) (1990), Römpp Chemie Lexikon, 9th edition, Vol. 3. Thieme, Stuttgart.
FARCASIU, D. (1975), J. Chem. Educ., 52, 76-79.
HAMMETT, L. P. (1940, 1970), "Physical Organic Chemistry", 1st and 2nd editions, McGraw Hill, New York.
HAMMOND, G. S. (1955), J. Am. Chem. Soc., 77, 334-338.
HANSCH, C., and LEO, A. J. (1979), "Substituent Constants for Correlation Analysis in Chemistry and Biology", Wiley, New York.
HO, T.-L. (1977), "Hard and Soft Acids and Bases Principle in Organic Chemistry", Academic Press, New York.
HOFMANN, A. W. (1851), Justus Liebigs Ann. Chem., 78, 253-286; 79, 11-39.
*IUPAC INORGANIC NOMENCLATURE (1990). IUPAC: Nomenclature of Inorganic Chemistry, Recommendations 1990, (LEIGH, G. J., Ed.), Blackwell, Oxford.
IUPAC NAMES FOR HYDROGEN ATOMS (1988). IUPAC: Organic Chemistry Division: Commission on Physical Organic Chemistry. Names for Hydrogen Atoms, Ions, and Groups, and for Reactions Involving Them. Pure Appl. Chem., 60, 1115-1116.
*IUPAC PHOTOCHEMICAL GLOSSARY (1988). IUPAC: Organic Chemistry Division: Commission on Photochemistry. Glossary of Terms Used in Photochemistry. Pure Appl. Chem., 60, 1055-1106.
KOHNSTAM, G. (1967), Adv. Phys. Org. Chem., 5, 121-172.
LEFFLER, J. E. (1953), Science, 117, 340-341.
PEARSON, R. G. (1963), J. Am. Chem. Soc., 85, 3533-3539.
PEARSON, R. G. (1973), "Introduction to Hard and Soft Acids and Bases". Dowden, Hutchinson, Ross, Stroudsburg.
RADOM, L. (1982), Progr. Theor. Org. Chem., 3, 1.
REICHARDT, C., ASHARIN-FARD, S., and SCHÄFER, G. (1993), Chem. Ber., 126, 143-147.
WILLIAMS, A. (1984), Acc. Chem. Res., 17, 425-430.
Continue with terms starting with I.
Return to home page for Glossary of terms used in Physical Organic Chemistry.