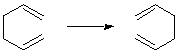
Contents
+ dative bond; decomposition, chemical; + de-electronation; degenerate chemical reaction; degenerate rearrangement; delocalization; deshielding; detachment; detailed balancing, principle of; diamagnetic; diastereoisomerism; dielectric constant; dienophile; diffusion-controlled rate; dimerization; Dimroth-Reichardt ET parameter; dipolar aprotic solvent; dipolar bond; dipolar cycloaddition; dipole-dipole interaction; dipole-induced dipole forces; diradical; direct effect; dismutation; dispersion forces; disproportionation; disrotatory; dissociation; distonic radical cation; donor number (DN); downfield; driving force (also called the affinity) of a reaction; dual substituent-parameter equation; dyotropic rearrangement
See coordination.
The breakdown of a single entity (normal molecule, reaction intermediate, etc.) into two or more fragments.
See oxidation (1).
See identity reaction.
A molecular rearrangement in which the principal product is indistinguishable (in the absence of isotopic labelling) from the principal reactant. The term includes both "degenerate intramolecular rearrangements" and reactions that involve intermolecular transfer of atoms or groups ("degenerate intermolecular rearrangements"): both are degenerate isomerizations. The occurrence of degenerate rearrangements may be detectable by isotopic labelling or by dynamic NMR techniques. For example: the [3,3]sigmatropic rearrangement of hexa-1,5-diene (Cope rearrangement),
Synonymous but less preferable terms are "automerization", "permutational isomerism", "isodynamic transformation", "topomerization". BINSCH, ELIEL, and KESSLER (1971). See also fluxional, molecular rearrangement, valence isomer.
A quantum mechanical concept most usually applied in organic chemistry to describe the pi bonding in a conjugated system. This bonding is not localized between two atoms: instead, each link has a "fractional double bond character" or bond order. There is a corresponding "delocalization energy", identifiable with the stabilization of the system compared with a hypothetical alternative in which formal (localized) single and double bonds are present. Some degree of delocalization is always present and can be estimated by quantum mechanical calculations. The effects are particularly evident in aromatic systems and in symmetrical molecular entities in which a lone pair of electrons or a vacant p-orbital is conjugated with a double bond (e.g. carboxylate ions, nitro compounds, enamines, the allyl cation). Delocalization in such species may be represented by partial bonds or as resonance (here symbolized by a two-headed arrow) between contributing structures.
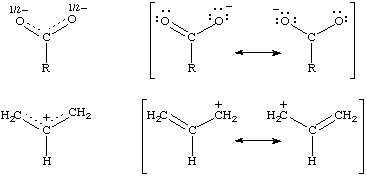
These examples also illustrate the concomitant delocalization of charge in ionic conjugated systems. Analogously, delocalization of the spin of an unpaired electron occurs in conjugated radicals. See also mesomerism.
See shielding.
The reverse of an attachment. See also electron attachment.
detailed balancing, principle of
When equilibrium is reached in a reaction system (containing an arbitrary number of components and reaction paths), as many atoms, in their respective molecular entities, will pass forward, as well as backwards, along each individual path in a given finite time interval. Accordingly, the reaction path in the reverse direction must in every detail be the reverse of the reaction path in the forward direction (provided always that the system is at equilibrium).
The principle of detailed balancing is a consequence for macroscopic systems of the principle of microscopic reversibility.
Substances having a negative magnetic susceptibility are diamagnetic. They are repelled out of a magnetic field. See also paramagnetic.
Stereoisomerism other than enantiomerism. Diastereoisomers (or diastereomers) are stereoisomers not related as mirror images. Diastereoisomers are characterized by differences in physical properties, and by some differences in chemical behaviour toward achiral as well as chiral reagents. IUPAC STEREOCHEMICAL TERMINOLOGY (1993).
A measure for the effect of a medium on the potential energy of interaction between two charges. It is measured by comparing the capacity of a capacitor with and without the sample present.
The olefin component of a Diels-Alder reaction. See cycloaddition.
See encounter-controlled rate, microscopic diffusion control. See also mixing control.
The transformation of a molecular entity A to give a molecular entity A2. For example:
2 CH3COCH3 ![]() (CH3)2C(OH)CH2COCH3
(CH3)2C(OH)CH2COCH3
2 RCOOH ![]() (RCOOH)2
(RCOOH)2
See also association.
Dimroth-Reichardt ET parameter
A measure of the ionizing power (loosely polarity) of a solvent, based on the maximum wavenumber of the longest wavelength electronic absorption band of
in a given solvent. ET, called ET(30) by its originators, is given by
![[nu]](../greek/NU.GIF)
![[lambda]](../greek/LAMBDA.GIF) -1 is in cm-1 and is in nm.
-1 is in cm-1 and is in nm.The so-called normalized ETN scale is defined as
DIMROTH, REICHARDT, SIEPMANN and BOHLMANN (1963); REICHARDT (1988). See also Grunwald-Winstein equation, Z-value.
A solvent with a comparatively high relative permittivity (or dielectric constant), greater than ca. 15, and a sizable permanent dipole moment, that cannot donate suitably labile hydrogen atoms to form strong hydrogen bonds, e.g. dimethyl sulfoxide. The term (and its alternative "polar aprotic solvent") is a misnomer and is therefore discouraged. Such solvents are usually not aprotic, but protophilic (and at most weakly protogenic). In describing a solvent it is better to be explicit about its essential properties, e.g. dipolar and non-protogenic.
A bond formed (actually or conceptually) by coordination of two neutral moieties, the combination of which results in charge-separated structures, e.g.,
The term is preferred to the obsolescent synonyms "coordinate link", "co-ordinate covalence", "dative bond", "semipolar bond".
See cycloaddition.
Intermolecular or intramolecular interaction between molecules or groups having a permanent electric dipole moment. The strength of the interaction depends on the distance and relative orientation of the dipoles. The term applies also to intramolecular interactions between bonds having permanent dipole moments. See also van der Waals forces.
See van der Waals forces.
See biradical.
See field effect.
See disproportionation.
See London forces.
Any chemical reaction of the type A + A ![]() A' + A", where A, A' and A" are different chemical species. For example:
A' + A", where A, A' and A" are different chemical species. For example:
The reverse of disproportionation is called comproportionation.
A special case of disproportionation (or "dismutation") is "radical disproportionation", exemplified by
Reactions of the more general type
are also loosely described as radical disproportionations.
(A somewhat more restricted usage of the term prevails in inorganic chemistry.)
(1) The separation of a molecular entity into two or more molecular entities (or any similar separation within a polyatomic molecular entity). Examples include unimolecular heterolysis and homolysis, and the separation of the constituents of an ion pair into free ions.
(2) The separation of the constituents of any aggregate of molecular entities.
In both senses dissociation is the reverse of association.
A radical cation in which charge and radical sites are separated. YATES, BOUMA and RADOM (1986)/a>.
A quantitative measure of Lewis basicity devised by GUTMANN (1976).
See chemical shift.
driving force (also called the affinity) of a reaction, A (SI unit: kJ mol-1)
The decrease in Gibbs energy on going from the reactants to the products of a chemical reaction (-
dual substituent-parameter equation
In a general sense, this is any equation which expresses substituent effects in terms of two parameters. However, in practice the term is used more specifically for an equation for summarizing the effects of meta- or para-substituents (i = m or p) X on chemical reactivity, spectroscopic properties, etc. of a probe site Y in benzene or other aromatic system.
P is the magnitude of the property Y for substituent X, expressed relative to the property for X=H;
An uncatalyzed process in which two sigma bonds simultaneously migrate intramolecularly, e.g.
BINSCH, G., ELIEL, E. L., and KESSLER, H. (1971), Angew. Chem., Int. Ed. Engl., 10, 570-572.
EHRENSON, S., BROWNLEE, R. T. C., and TAFT, R. W. (1973), Progr. Phys. Org. Chem., 10, 1-80.
GUTMANN, V. (1976), Coord. Chem. Rev., 18, 225-255.
*IUPAC STEREOCHEMICAL TERMINOLOGY (1993). IUPAC: Organic Chemistry Division: Basic Terminology of Stereochemistry. IDCNS and public review. Now published as Basic Terminology of Stereochemistry (IUPAC Recommendations 1996) in Pure Appl. Chem., 68, 2193-2222 (1996).
REETZ, M. (1972), Angew. Chem., Int. Ed. Engl., 11, 129-130, 130-131.
TAFT, R. W., Jr., and TOPSOM, R. D. (1987), Progr. Phys. Org. Chem., 16, 1-83.
YATES, B. F., BOUMA, W. J., and RADOM, L. (1986), Tetrahedron, 42, 6225-6234.
![[Delta]](../greek/DDELTA.GIF) G).
G).![[rho]](../greek/RHO.GIF) Ii
Ii![[sigma]](../greek/SIGMA.GIF) I + RiRI and R are inductive or polar and resonance substituent constants, respectively, there being various scales for R; I and R are the corresponding regression coefficients. EHRENSON, BROWNLEE, and TAFT (1973); TAFT and TOPSOM (1987). See also extended Hammett equation.
I + RiRI and R are inductive or polar and resonance substituent constants, respectively, there being various scales for R; I and R are the corresponding regression coefficients. EHRENSON, BROWNLEE, and TAFT (1973); TAFT and TOPSOM (1987). See also extended Hammett equation.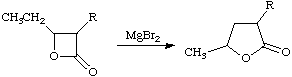
References
Continue with terms starting with E.
Return to home page for Glossary of terms used in Physical Organic Chemistry.